
Download
SHORT COMMUNICATION
Gastrointestinal stenosis: an underrecognized complication of CARMIL2 deficiency
Khaoula Oussamaa*, Ibtihal Benhsaiena,b, Vivien Béziatc,d,e, Halima Msaafb, Ikram Fetnassib, Jalila El bakkouria,f, Mounia Alzemmourig, Ahmed Aziz Bousfihaa,b, Zineb Hammoumig, Fatima Ailala,b
aLaboratory of Clinical Immunology, Infection and Autoimmunity LICIA, Faculty of Medicine and Pharmacy, Hassan II University, Casablanca, Morocco
bClinical Immunology and Infectious Pediatrics Department, Abderrahim Harouchi Hospital, Ibn Rochd University Hospital, Casablanca, Morocco
cLaboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM, Necker Hospital for Sick Children, Paris, France
dImagine Institute, University of Paris-Cité, Paris, France
eSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA
fImmunology Laboratory, Ibn Rochd University Hospital, Casablanca, Morocco
gDepartment of Pediatric Surgery, Abderrahim Harouchi Hospital, Ibn Rochd University Hospital, Casablanca, Morocco
Abstract
CARMIL2 deficiency is a rare autosomal recessive combined immunodeficiency classically associated with dermatitis, inflammatory bowel disease (IBD), recurrent infections, and Epstein–Barr virus-related tumors. Gastrointestinal (GI) stenosis remains an underrecognized but potentially life-threatening complication. We report a consanguineous Moroccan family in which all three siblings with CARMIL2 deficiency developed early-onset, severe, and progressive GI stenoses, including pyloric and esophageal involvement. The striking consistency and severity of this phenotype within a single family highlight the clinical importance of early recognition and timely intervention to prevent irreversible GI damage.
Key words: CARMIL2 deficiency, primary immunodeficiency, gastrointestinal stenosis, clinical vigilance
*Corresponding author: Khaoula Oussama, Laboratory of Clinical Immunology, Infection and Autoimmunity, LICIA, Faculty of Medicine and Pharmacy, Hassan II University, Casablanca, Morocco. Email address: [email protected]
Received: 6 February 2026; Accepted 20 February 2026; Available online: 1 May 2026
Copyright: Oussama K, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
CARMIL2 deficiency, caused by biallelic loss-of-function mutations in the CARMIL2 gene, results in a combined immunodeficiency characterized by recurrent infections, dermatitis, inflammatory bowel disease (IBD), warts, abscesses, chronic mucocutaneous candidiasis, and Epstein–Barr virus (EBV)–associated smooth muscle tumors.1
Many studies have highlighted the critical role of CARMIL2 in the development of memory B cells, regulatory T cells (Tregs) and natural killer (NK) cells.2–4 Moreover, CARMIL2 is essential for the maturation of naive CD4+ T cells into memory T cells, as well as their differentiation into T helper 1 (Th1), T helper 17 (Th17), and T follicular helper (Tfh) subsets.4
Levy et al recently demonstrated that CD28 signaling deficiency cannot explain all immunological and clinical phenotype in patients with CARMIL2 deficiency.2 Indeed, patients with CD28 deficiency develop an exquisite susceptibility to skin papillomavirus infection, but lack all other salient clinical features of CARMIL2 deficiency.2,5 In addition, CD28 deficient patients have a mild excess of naïve T cells, and an important decrease of Tregs, but their other leukocyte subsets are otherwise normal. These data, coupled to the broad expression of CARMIL2 in lymphocytes, suggest that CARMIL2 plays a key role beyond CD28 signaling inside T cells, but also downstream unknown receptors inside B cells and NK cells. One hypothesis is that CARMIL2 is crucial for PKC-dependent activation of NF-κB downstream CARD11.2 In line with such hypothesis, CARMIL2 deficient B cells fail to activate NF-κB-pathway upon IgM activation, leading to impaired B cell responses and bacterial respiratory infections in the patients(2,4). In addition, impaired cytoskeleton organization probably contributes to pathogenesis in CARMIL2 deficient individual.3,6,7
Since its initial description, CARMIL2 deficiency—also referred to as RLTPR deficiency—has been increasingly associated with gastrointestinal (GI) manifestations, predominantly of inflammatory origin.8 These include colitis, Crohn’s disease–like phenotypes, GI ulcerations, and eosinophilic esophagitis, often leading to chronic diarrhea, abdominal pain, malabsorption, and growth failure. In some patients, prolonged inflammation may culminate in fibrotic remodeling and GI stenosis, frequently refractory to medical therapy and requiring endoscopic or surgical intervention.3,8,9 To date, only a limited number of cases of GI strictures have been reported in association with CARMIL2 deficiency,2,8–11 suggesting that this complication remains underrecognized.
Materials and Methods
Ethics statement
This study was conducted in accordance with the principles of the Declaration of Helsinki. Approval was obtained from the Ethics Committee of CHU Ibn Rochd, Casablanca, Morocco. Written informed consent for participation and publication was obtained from all patients and/or their legal guardians.
Participants
Three affected siblings of Moroccan ancestry were recruited from Pediatric Unit 1 at the Ibn Rochd University Hospital Center. In addition, both parents and one unaffected sister were enrolled for genetic segregation analysis. The diagnosis of affected individuals was based on clinical presentation and confirmed by genetic analysis.
Genetic analysis
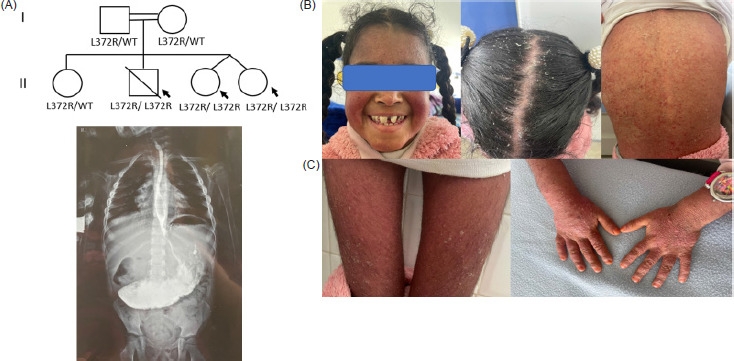
Genomic DNA was extracted from peripheral blood samples using standard procedures. Whole-exome sequencing (WES) was performed to identify causative variants. Bioinformatic analysis focused on rare homozygous variants consistent with an autosomal recessive inheritance pattern.The homozygous CARMIL2 p.Leu372Arg variant was identified in all three affected siblings. Segregation analysis was performed by Sanger sequencing in available family members. Both parents and the unaffected sister were heterozygous carriers, while all affected siblings were homozygous, consistent with an autosomal recessive mode of inheritance (Figure 1A).
Figure 1 (A) Pedigree of the family. (B) One of the twins (Patient II.3) exhibited severe psoriasis involving the entire body, including the lower and upper limbs, face, trunk, and scalp. (C) Upper gastrointestinal contrast study of patient II.3, revealing massive gastric stasis consistent with hypertrophic pyloric stenosis.
Phenotypic analysis
Clinical records of the three affected siblings were retrospectively reviewed. Data collected included family and medical history, clinical manifestations, hospitalizations, and treatments. Phenotypic characterization focused on immune-related features and other relevant clinical complications associated with CARMIL2 deficiency.
Results
We report a consanguineous Moroccan family with three affected siblings (Figure 1A): two surviving twin sisters and a brother who died at 17 years due to respiratory complications. The first twin (Patient II.3) presented at 18 months with a cytomegalovirus (CMV) respiratory infection complicated by localized bronchiectasis and recurrent infections. She also exhibited toenail onychomycosis. At 2 years, generalized psoriasiform lesions developed on the face, trunk, and limbs (Figure 1B). Recurrent respiratory infections persisted throughout childhood, primarily caused by CMV, Haemophilus influenzae, and Streptococcus pneumoniae. GI manifestations included intermittent diarrhea, infections, and recurrent oral thrush. At 8 years, she developed pyloric stenosis, presenting as persistent postprandial vomiting and epigastric bloating. Radiographic evaluation revealed severe gastric stasis and pyloric narrowing, and she underwent a Ramstedt pyloromyotomy with complete resolution of symptoms (Figure 1C). Laboratory findings showed severe anemia, leukocytosis with lymphocytosis, neutrophilia, monocytosis, thrombocytosis, and elevated T and B lymphocyte counts (Table 1). Humoral assessment revealed moderate hypogammaglobulinemia with slightly elevated IgM and decreased IgA and IgG, while endocrine evaluation showed reduced IGF-1 levels.
Table 1 Hematological, immunological, and hormonal profiles of three siblings with CARMIL2 deficiency.
| Parameter | Patient II.3 | Patient II.4 | Patient II.2 | Normal range |
|---|---|---|---|---|
| Hematological parameters | ||||
| Hemoglobin (Hb) | 7.2 g/dL | 7.7 g/dL | 6.7 g/dL | 12–14.5 g/dL |
| White blood cells | 20,100/mm3 | 8260/mm3 | 20,060/mm3 | 4500–11,000/mm3 |
| Absolute lymphocyte count (CBC) | 8522/mm3 | 3580/mm3 | 4193/mm3 | 2000–5000/mm3 |
| Neutrophils | 9548/mm3 | 3890/mm3 | 14,483/mm3 | 2000–6000/mm3 |
| Eosinophils | 643/mm3 | 210/mm3 | 60/mm3 | 100–600/mm3 |
| Basophils | 40/mm3 | 30/mm3 | 40/mm3 | 0–150/mm3 |
| Monocytes | 1347/mm3 | 550/mm3 | 1284/mm3 | 200–800/mm3 |
| Platelets | 827,000/mm3 | 349,000/mm3 | 821,000/mm3 | 160,000–450,000/mm3 |
| Lymphocyte subsets (flow cytometry) | ||||
| CD3+T cells | 5553/mm3 | 6222/mm3 | 4074/mm3 | 500–1900/mm3 |
| CD3+CD4+T cells | 3108/mm3 | 3095/mm3 | 1292/mm3 | 300–1300/mm3 |
| CD3+CD8+T cells | 2117/mm3 | 2555/mm3 | 1888/mm3 | 200–1000/mm3 |
| CD19+B cells | 1566/mm3 | 1314/mm3 | 546/mm3 | 80–490/mm3 |
| CD16+CD56+NK cells | 807/mm3 | 434/mm3 | 248/mm3 | 60–495/mm3 |
| Immunoglobulin profile | ||||
| IgA | 0.72 g/L | 0.80 g/L | 0.93 g/L | 0.8–4.0 g/L |
| IgG | 4.40 g/L | 8.20 g/L | 8.55 g/L | 5.04–14.64 g/L |
| IgM | 2.54 g/L | 2.70 g/L | 2.09 g/L | 0.4–2.3 g/L |
| IgE | <5 kUI/L | <5 kUI/L | <5 kUI/L | <114 kUI/L |
| Hormonal profile | ||||
| IGF-1 | 63.62 ng/mL | 51.75 ng/mL | – | 80–244 ng/mL |
Her twin sister (Patient II.4) exhibited a similar early course, with CMV respiratory infection complicated by localized bronchiectasis and recurrent infections, toenail onychomycosis, and intermittent GI symptoms. She developed mild erythroderma at 3 years, which was less severe than her sister’s lesions. Pyloric stenosis occurred at 6 years and was surgically corrected, resulting in complete resolution. Laboratory evaluation revealed anemia, normal total leukocyte and platelet counts, elevated T and B lymphocytes, and increased IgM, with normal IgA, IgG, and IgE (Table 1).
The twins initially received intravenous immunoglobulin therapy; however, this treatment was poorly tolerated. As a result, anti-infective prophylaxis with trimethoprim–sulfamethoxazole was subsequently initiated (Table 2).
Table 2 Clinical phenotype and outcomes of three siblings with CARMIL2 deficiency.
| Patients | Patient II.3 | Patient II.4 | Patient II.2 |
|---|---|---|---|
| Age at onset of first symptoms | 18 months | 18 months | 2 years |
| Clinical manifestations | Recurrent respiratory infections, recurrent gastrointestinal infections, severe psoriasis, onychomycosis, pyloric stenosis | Recurrent respiratory infections, recurrent gastrointestinal infections, mild erythroderma, onychomycosis, pyloric stenosis | Recurrent respiratory infections, erythrodermic psoriasis, infected radial fracture of the elbow, multifocal tuberculosis, meningoencephalitis, recurrent gastrointestinal infections, onychomycosis, esophageal stenosis |
| Treatment | Antibiotics (amoxicillin, trimethoprim–sulfamethoxazole), antiviral therapy (ganciclovir), methotrexate, folic acid | Antibiotics (amoxicillin, trimethoprim–sulfamethoxazole), antiviral therapy (ganciclovir) | Corticosteroids, intravenous antibiotics,anti-tuberculosis therapy, empirical antibiotic and antiviral therapy, pneumatic dilation, diuretics, ACE inhibitors, digoxin, antiplatelet therapy |
| Outcome | Alive | Alive | Deceased |
The male sibling (Patient II.2) had a more severe course, beginning in early childhood with failure to thrive, recurrent infections, and erythrodermic psoriasis. He developed multifocal tuberculosis at 8 years and meningoencephalitis at 9 years. Immunological assessment revealed iron-deficiency anemia, leukocytosis with neutrophilia, thrombocytosis, and elevated T lymphocytes, with preserved humoral immunity (Table 1). At 16 years, he developed progressive dysphagia due to esophageal stricture with associated esophagitis and nodular pangastritis, partially relieved by pneumatic dilation. Later, he presented with respiratory distress, decompensated cardiomyopathy, pulmonary hypertension, and adrenal and renal masses suggestive of catecholamine-producing tumors. Despite multidisciplinary care, he died at 17 years from severe respiratory complications (Table 2).
Discussion
Although CARMIL2 deficiency is rare, GI stenosis appears to represent a clinically meaningful complication. Including the present family, approximately 13% of reported CARMIL2-deficient patients (12/89) have developed GI stenoses, underscoring the clinical relevance of this complication.2
Several studies have reported pathogenic CARMIL2 mutations associated with GI stenoses. Lévy et al. described six new cases from five families, all presenting with GI stenoses linked to CARMIL2 mutations. The variants included different mutation types: a synonymous variant affecting splicing (c.1578C>T), a frameshift mutation (c.1906_1907del; p.L636Afs39), a nonsense mutation (c.790C>T; p.R264), a splice-site mutation (c.1226+1G>T), and a missense mutation (c.1825G>A; p.D609N).2 This heterogeneity suggests that diverse molecular alterations, regardless of type, can converge toward a severe clinical phenotype, particularly affecting the GI tract.
For example, Magg et al. reported two pairs of Turkish siblings carrying a homozygous frameshift deletion (c.688_689delAG; p.S230Pfs*2). Immunological profiling of one sibling revealed decreased regulatory T cells (Tregs), central memory T cells (TCM), and effector memory T cells (TEM), along with increased naïve B cells and decreased marginal zone B cells (BMZ) and switched memory B cells. Both siblings developed pyloric stenosis in early life, requiring surgical intervention.8 Additionally, Alazami et al.9 reported a Saudi Arabian female with a frameshift mutation (c.2536_2548del; p.Leu846Serfs), who developed esophageal stenosis. Her immunological evaluation showed a significant decrease in CD4+ memory T cells and total memory T cells, with elevated naïve T cells. Marangi et al.10 described an Italian girl with a nonsense mutation (c.1109C>A; p.Ser370Ter) who presented with esophageal stenosis, reduced Treg numbers, and diminished memory B cells, although Th1 and Th17 subsets were within normal ranges. Sorte et al.11 also reported a female patient with recurrent GI stenoses carrying a missense mutation (c.1808T>A; p.L603H).
The present report is distinctive for documenting a consistent, severe stenotic phenotype affecting all affected siblings within a single family.
According to the literature, CARMIL2 deficiency is associated with a wide range of GI manifestations, most commonly IBD, chronic diarrhea, and recurrent abdominal pain. Several patients have been diagnosed with Crohn’s disease or have presented with severe inflammatory phenotypes, including pancolitis or colitis, as well as dysphagia. Less frequent involvement of other segments of the GI tract has also been reported, such as gastritis, duodenitis, various forms of esophagitis, bloody diarrhea, and eosinophilic gastroenteritis. Rare and atypical manifestations—including Candida esophagitis, necrotizing enterocolitis, esophageal webs, celiac disease, or crypt abscess formation—have also been described.1–3,8–21
In general, GI stenosis may arise as a late complication of chronic inflammatory disease, particularly in settings characterized by persistent mucosal inflammation, recurrent ulceration, or prolonged tissue injury. Such processes are known in other inflammatory GI disorders to result in luminal narrowing through complex, heterogeneous mechanisms that may include edema, muscular hypertrophy, or long-term tissue remodeling. However, the relative contributions of these mechanisms vary across diseases and remain incompletely understood.
In the context of CARMIL2 deficiency, several GI manifestations reported in the literature occur in inflammatory settings, where stenosis can develop. Nevertheless, the pathophysiological basis of stenosis formation in this condition remains unclear. While repeated mucosal injury and chronic inflammation could, in theory, promote progressive tissue remodeling, there is currently no direct evidence of fibrotic remodeling in patients with CARMIL2 deficiency.
In the affected siblings reported here, the severe GI stenoses are therefore most plausibly interpreted as a consequence of long-standing inflammatory involvement of the digestive tract. Histopathological confirmation of fibrosis is not available, and imaging findings are nonspecific and do not permit direct assessment of fibrotic changes. Accordingly, fibrosis cannot be inferred and is mentioned only as a speculative hypothesis, based on the clinical course rather than on objective pathological evidence. This interpretation should be regarded as cautious and hypothesis-generating, rather than mechanistically conclusive.
CARMIL2 deficiency may predispose patients to severe GI complications as a consequence of persistent immune dysregulation and chronic intestinal inflammation. From a clinical perspective, this underscores the importance of recognizing atypical or progressive digestive manifestations, considering early immunomodulatory strategies to limit the progression toward stenotic disease, and promoting collaborative multicenter efforts to refine management approaches. Moreover, further investigation into the role of CARMIL2 in intestinal barrier integrity could open new avenues for targeted therapeutic interventions.
In conclusion, this report identifies severe GI stenosis as a clinically significant and likely underappreciated feature of CARMIL2 deficiency, particularly when observed in a recurrent familial context. The consistent pattern described here supports the need for proactive diagnostic assessment and individualized management strategies to mitigate long-term GI morbidity.
Acknowledgments
We are deeply grateful to the patients and their families for their kindness, trust, and willingness to contribute to this study. Their generosity made this work possible.We also thank Professor Jean-Laurent Casanova for his valuable scientific input and insightful discussions.
Mandatory Disclosure on Use of Artificial Intelligence
The authors declare that AI-assisted tools were used for minor language editing only. All references have been manually verified for accuracy and relevance.
Author Contributions
All authors contributed equally to this article.
Conflict of Interest
The authors declare no conflicts of interest.
Funding
None.
REFERENCES
1 Kolukisa B, Baser D, Akcam B, Danielson J, Bilgic Eltan S, Haliloglu Y, et al. Evolution and long-term outcomes of combined immunodeficiency due to CARMIL2 deficiency. Allergy. 2022 Mar;77(3):1004–19. 10.1111/all.15010
2 Lévy R, Gothe F, Momenilandi M, Magg T, Materna M, Peters P, et al. Human CARMIL2 deficiency underlies a broader immunological and clinical phenotype than CD28 deficiency. J Exp Med. 2023 Feb 6;220(2):e20220275. 10.1084/jem.20220275
3 Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun. 2017 Jan 23;8:14209. 10.1038/ncomms14209
4 Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell-and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016 Oct 17;213(11):2413–35. 10.1084/jem.20160576
5 Béziat V, Rapaport F, Hu J, Titeux M, Bonnet des Claustres M, Bourgey M, et al. Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell. 2021 Jul 8;184(14):3812–3828.e30. 10.1016/j.cell.2021.06.004
6 Lanier MH, Kim T, Cooper JA. CARMIL2 is a novel molecular connection between vimentin and actin essential for cell migration and invadopodia formation. Mol Biol Cell. 2015 Dec 15;26(25):4577–88. 10.1091/mbc.E15-08-0552
7 Lanier MH, McConnell P, Cooper JA. Cell Migration and Invadopodia Formation Require a Membrane-binding Domain of CARMIL2. J Biol Chem. 2016 Jan 15;291(3):1076–91. 10.1074/jbc.M115.676882
8 Magg T, Shcherbina A, Arslan D, Desai MM, Wall S, Mitsialis V, et al. CARMIL2 Deficiency Presenting as Very Early Onset Inflammatory Bowel Disease. Inflamm Bowel Dis. 2019 Oct 18;25(11):1788–95. 10.1093/ibd/izz103
9 Alazami AM, Al-Helale M, Alhissi S, Al-Saud B, Alajlan H, Monies D, et al. Novel CARMIL2 Mutations in Patients with Variable Clinical Dermatitis, Infections, and Combined Immunodeficiency. Front Immunol. 2018;9:203. 10.3389/fimmu.2018.00203
10 Marangi G, Garcovich S, Sante GD, Orteschi D, Frangella S, Scaldaferri F, et al. Complex Muco-cutaneous Manifestations of CARMIL2-associated Combined Immunodeficiency: A Novel Presentation of Dysfunctional Epithelial Barriers. Acta Derm Venereol. 2020 Jan 23;100(1):adv00038. 10.2340/00015555-3370
11 Sorte HS, Osnes LT, Fevang B, Aukrust P, Erichsen HC, Backe PH, et al. A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Mol Genet Genomic Med. 2016 Nov;4(6):604–16. 10.1002/mgg3.237
12 Bosa L, Batura V, Colavito D, Fiedler K, Gaio P, Guo C, et al. Novel CARMIL2 loss-of-function variants are associated with pediatric inflammatory bowel disease. Sci Rep. 2021 Mar 15;11(1):5945. 10.1038/s41598-021-85399-9
13 Chan A, Feuille E, Bassetti J, Pereira E, Demirdag Y. M232 Variable Phenotypes Associated With P.H648tfsx20 Pathogenic Variant In Carmil2 Gene: A Report Of Two Cases. Annals of Allergy, Asthma & Immunology. 2019 Nov 1;123(5):S104. 10.1016/j.anai.2019.08.204
14 Frede N, Rojas-Restrepo J, Caballero Garcia de Oteyza A, Buchta M, Hübscher K, Gámez-Díaz L, et al. Genetic Analysis of a Cohort of 275 Patients with Hyper-IgE Syndromes and/or Chronic Mucocutaneous Candidiasis. J Clin Immunol. 2021 Nov;41(8):1804–38. 10.1007/s10875-021-01086-4
15 Kim D, Uner A, Saglam A, Chadburn A, Crane GM. Peripheral eosinophilia in primary immunodeficiencies of actin dysregulation: A case series of Wiskott-Aldrich syndrome, CARMIL2 and DOCK8 deficiency and review of the literature. Ann Diagn Pathol. 2019 Dec;43:151413. 10.1016/j.anndiagpath.2019.151413
16 Kurolap A, Eshach Adiv O, Konnikova L, Werner L, Gonzaga-Jauregui C, Steinberg M, et al. A Unique Presentation of Infantile-Onset Colitis and Eosinophilic Disease without Recurrent Infections Resulting from a Novel Homozygous CARMIL2 Variant. J Clin Immunol. 2019 May;39(4):430–9. 10.1007/s10875-019-00631-6
17 Maccari ME, Speckmann C, Heeg M, Reimer A, Casetti F, Has C, et al. Profound immunodeficiency with severe skin disease explained by concomitant novel CARMIL2 and PLEC1 loss-of-function mutations. Clin Immunol. 2019 Nov;208:108228. 10.1016/j.clim.2019.06.004
18 Rao LP, Kothiwale V, Radhakrishnan P, Rangaswamy D, Shukla A, Bhat V. Secondary Membranous Nephropathy and Immunodeficiency due to a Novel Biallelic Variant in CARMIL2. Indian J Nephrol. 2024;34(6):667–9. 10.25259/ijn_542_23
19 Shamriz O, Simon AJ, Lev A, Megged O, Ledder O, Picard E, et al. Exogenous interleukin-2 can rescue in-vitro T cell activation and proliferation in patients with a novel capping protein regulator and myosin 1 linker 2 mutation. Clin Exp Immunol. 2020 Jun;200(3):215–27. 10.1111/cei.13432
20 Shayegan LH, Garzon MC, Morel KD, Borlack R, Vuguin PM, Margolis KG, et al. CARMIL2-related immunodeficiency manifesting with photosensitivity. Pediatr Dermatol. 2020 Jul;37(4):695–7. 10.1111/pde.14173
21 Yonkof JR, Gupta A, Rueda CM, Mangray S, Prince BT, Rangarajan HG, et al. A Novel Pathogenic Variant in CARMIL2 (RLTPR) Causing CARMIL2 Deficiency and EBV-Associated Smooth Muscle Tumors. Front Immunol. 2020;11:884. 10.3389/fimmu.2020.00884