
Download
ORIGINAL ARTICLE
Amygdalin attenuates interleukin-1β-stimulated chondrocyte damage in mice through the Nrf2/NF-κB pathway
Li Zhanga, Yan Lib*
aDepartment of Orthopedics, Wuhan Hankou Hospital, Wuhan, Hubei Province, China
bDepartment of Respiratory Medicine, Wuhan Hankou Hospital, Wuhan, Hubei Province, China
Abstract
Background: Osteoarthritis (OA) is the most usual joint disease, which affects the life of patients and causes them seriously physical pain. Amygdalin, the main active pharmaceutical ingredient in Semen Armeniacae Amarum, has anti-inflammatory, anti-oxidation, and immunomodulatory impacts. However, the regulatory functions of Amygdalin in OA progression keep indistinct and need further investigations. This study is aimed to probe the regulatory influences and associated pathways of Amygdalin in OA development.
Methods: The cell viability was confirmed through CCK-8 assay. The levels of PGE2, TNF-α and IL-6 were detected through ELISA. The ROS level was examined through DCF staining. The levels of MDA, SOD and GSH were measured through the commercial kit. The protein expressions were evaluated through western blot.
Results: Firstly, it was demonstrated that Amygdalin can refrain IL-1β-evoked inflammation in chondrocytes. Next, Amygdalin repressed IL-1β-triggered oxidative stress in chondrocytes. Moreover, Amygdalin inhibited IL-1β-mediated ECM degradation in chondrocytes. Lastly, it was revealed that Amygdalin accelerated the Nrf2 pathway and suppressed the NF-κB pathway.
Conclusion: Amygdalin attenuated IL-1β-stimulated chondrocyte damage through the Nrf2/NF-κB pathway, thereby ameliorating OA progression. This finding hinted that Amygdalin may be one promising drug for OA treatment.
Key words: amygdalin, chondrocyte damage, Nrf2/NF-κB pathway, osteoarthritis
*Corresponding author: Yan Li, Department of Respiratory Medicine, Wuhan Hankou Hospital, No. 7 Erqi Side Road, Jiang’an District, Wuhan City, Hubei Province 430012, China. Email address: [email protected]
Received 31 July 2025; Accepted 29 September 2025; Available online 1 January 2026
Copyright: Zhang L and Yan L
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Osteoarthritis (OA) is the most prevalent degenerative joint disorder affecting approximately 500 million adults globally and resulting in significant joint damage and intense pain.1,2 It primarily involves degradation of the articular hyaline cartilage in weight-bearing joints.3 As OA progresses, it is often accompanied by extensive cartilage destruction, subchondral bone thickening, joint space narrowing, osteophyte formation, and varying degrees of joint inflammation and pain.4 Current treatment strategies for OA are largely confined to symptomatic relief through nonsteroidal anti-inflammatory drugs or, in advanced cases, surgical joint replacement.5 Given the limitations of the existing therapies, the development of pharmacological agents capable of alleviating chondrocyte injury is of increasing importance in OA management.
Amygdalin, the principal bioactive compound extracted from Armeniacae semen amarum, has been shown to exert anti-inflammatory, antioxidant, and immunomodulatory effects.6 Emerging evidence suggests that amygdalin may be involved in modulating the progression of several pathological conditions. For instance, amygdalin has been reported to mitigate multidrug-resistant Staphylococcus aureus-induced lung epithelial cell injury by regulating inflammation and oxidative stress.7 In the context of intervertebral disc degeneration, amygdalin inhibits nuclear factor kappa B (NF-κB) signaling pathway and inflammatory responses to attenuate cartilage endplate degeneration.8 Besides, amygdalin can modulate Janus kinase 2–signal transducer and activator of transcription 3 (JAK2/STAT3) pathway to relieve inflammation and improve alopecia areata.9 In diabetic retinopathy, it reduces oxidative stress and ferroptosis by acting on the nuclear factor-erythroid 2-related factor-2–antioxidant response element (NRF2/ARE) signaling pathway.10 In addition, amygdalin suppresses osteoclast differentiation, endoplasmic reticulumstress, and reactive oxygen species (ROS) generation,11 indicating its potential as a natural therapeutic candidate for bone-related disorders. However, the specific regulatory effects of amygdalin and its molecular mechanisms in OA pathogenesis remain largely undefined.
In this study, we report for the first time that amygdalin can attenuate interleukin (IL)-1β-induced chondrocyte damage by modulating the Nrf2/NF-κB signaling pathway, thereby mitigating the progression of OA. These findings provide new insight into the therapeutic potential of amygdalin for treating OA.
Materials and Methods
Cell lines and treatment
Chondrocytes were isolated from the knee joint cartilage of 14-day-old C57BL/6J mice (Vital River Laboratory Animal Technology Co. Ltd., Beijing, China). The cartilage tissues were then dissected and digested with 0.25% trypsin at 37°C for 30 min, followed by further digestion with 0.1% collagenase at 37°C for 2 h. The digested samples were centrifuged, and the collected cells were cultured in Dulbecco’s modified eagle medium–nutrient mixture F-12 (DMEM/F12) medium supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2. To establish an in vitro OA model, the chondrocytes were treated with IL-1β (10 ng/mL) for 24 h. Amygdalin (5, 10, 20, 40, and 80 µM; Shanghai Selleck Chemicals Co. Ltd., Shanghai, China) was applied to the chondrocytes to assess its effects.
Cell counting kit-8 (CCK-8) assay
Chondrocytes were seeded into 96-well plates at a density of 1000 cells per well. Cell viability was evaluated using the CCK-8 (Dojindo Laboratories, Kumamoto, Japan). CCK-8 solution, 10 µL, was added to each well, and the absorbance was measured using a microplate reader (Bio-Rad, CA, USA).
Enzyme-linked immunosorbent serologic assay (ELISA)
The levels of tumor necrosis factor-α (TNF-α; ab208348), prostaglandin E2 (PGE2; ab287802), and interleukin-6 (IL-6; ab222503) in culture supernatants were quantified using commercially available ELISA kits (Abcam, Shanghai, China), according to the manufacturers’ protocols.
Detection of ROS (DCFH-DA staining)
The ROS generation was assessed using 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) staining (E004-1-1; Nanjing Jiancheng Technology Co. Ltd. Nanjing, China). Chondrocytes were incubated with DCFH-DA and washed subsequently. Fluorescence was observed and captured using a fluorescence microscope (Olympus, Tokyo, Japan).
Detection of MDA, SOD and GSH
Oxidative stress markers, such as malondialdehyde (MDA; ab118970), superoxide dismutase (SOD; ab65354), and glutathione (GSH; ab65322), were quantified using corresponding detection kits (Abcam, Shanghai, China) following manufacturers’ instructions.
Western blot analysis
Total proteins were extracted from chondrocytes using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China), separated via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking, the membranes were incubated with the following primary antibodies: anti-collagen II (ab34712, 1:1000), anti-MMP-13 (ab315267, 1:1000), anti-ADAMTS-5 (ab41037, 1:250), anti-phospho-p65 (ab76302, 1:1000), anti-p65 (ab32536, 1:1000), anti-phospho-IκBα (ab92700, 1:1000), anti-IκBα (ab32518, 1:1000), and anti-β-actin (ab8226, 1 µg/mL) (Abcam, Shanghai, China). After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (ab6721,1:2000). Protein bands were visualized using an enhanced chemiluminescence (ECL) kit (Santa Cruz Biotechnology, TX, USA).
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 9.0 (GraphPad Software, La Jolla, CA, USA). Data were expressed as mean ± standard deviation (SD); N = 3 repetitions. One-way analysis of variance (ANOVA) was used to evaluate statistical significance among groups; P < 0.05 was considered statistically significant.
Results
Amygdalin alleviated IL-1β-induced inflammation in chondrocytes
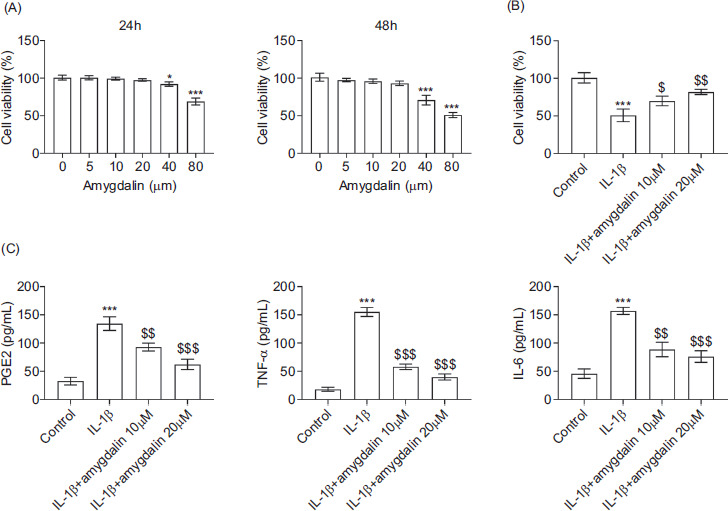
As shown in Figure 1A, cell viability was significantly reduced following amygdalin treatment at a concentration of 40 μM or 80 μM for 24 h and 48 h, indicating cytotoxicity at these doses. In contrast, treatment with 5, 10, or 20 μM did not result in cytotoxic effects, and these concentrations were selected for subsequent experiments. Notably, amygdalin treatment at 10 μM or 20 μM effectively restored the IL-1β-induced reduction in chondrocyte viability (Figure 1B). Furthermore, IL-1β stimulation markedly increased the secretion of proinflammatory mediators, such as prostaglandin E2 (PGE2), TNF-α, and IL-6; however, these elevations were significantly attenuated by amygdalin treatment at 10 μM or 20 μM (Figure 1C). Collectively, these findings demonstrate that amygdalin alleviates IL-1β-induced inflammatory responses in chondrocytes.
Figure 1 Amygdalin attenuates IL-1β-induced inflammation in chondrocytes. (A) Cell viability was assessed by CCK-8 assay in chondrocytes treated with 0-, 5-, 10-, 20-, 40-, or 80-μM amygdalin for 24 h. (B) Cell viability was measured by CCK-8 assay in the control, IL-1β, IL-1β+amygdalin (10 μM), and IL-1β+amygdalin (20 μM) groups. (C) Levels of PGE2, TNF-α, and IL-6 in the culture supernatant were quantified by ELISA. N = 3 repetitions.*P < 0.05, ***P < 0.001 vs the control group; $P < 0.05, $$P < 0.01, $$$p < 0.001 vs the IL-1β group.
Amygdalin suppressed IL-1β-induced oxidative stress in chondrocytes
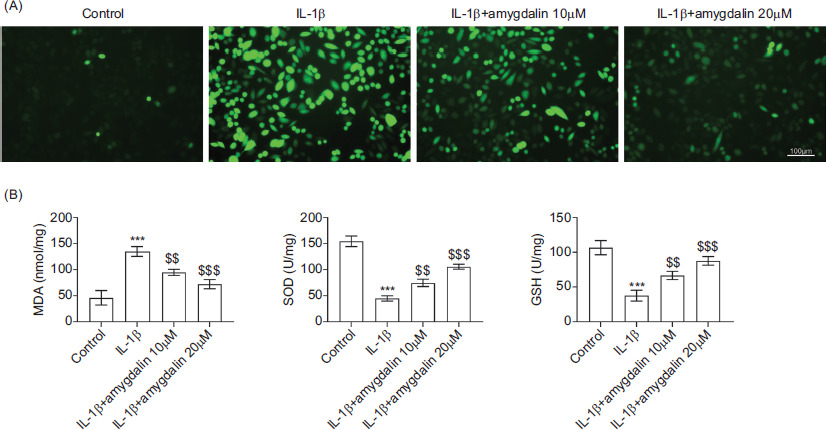
Intracellular ROS levels were assessed to determine whether amygdalin modulated oxidative stress. As illustrated in Figure 2A, IL-1β stimulation led to a marked increase in ROS production, which was significantly reduced by amygdalin treatment at 10 μM or 20 μM. Consistently, IL-1β treatment elevated MDA levels while suppressing SOD and GSH levels, indicative of oxidative damage. These alterations were largely reversed following amygdalin treatment (Figure 2B). These data suggest that amygdalin suppresses IL-1β-induced oxidative stress in chondrocytes.
Figure 2 Amygdalin suppresses IL-1β-induced oxidative stress in chondrocytes. Chondrocytes were divided into control, IL-1β, IL-1β+amygdalin (10 μM), and IL-1β+amygdalin (20 μM) groups. (A) Intracellular ROS levels were evaluated by DCFH-DA staining. (B) Levels of MDA, SOD, and GSH were measured using commercial assay kits. N = 3 repetitions. ***P < 0.001 vs the control group; $$P < 0.01, $$$P < 0.001 vs the IL-1β group.
Amygdalin inhibited IL-1β-mediated extracellular matrix (ECM) degradation in chondrocytes
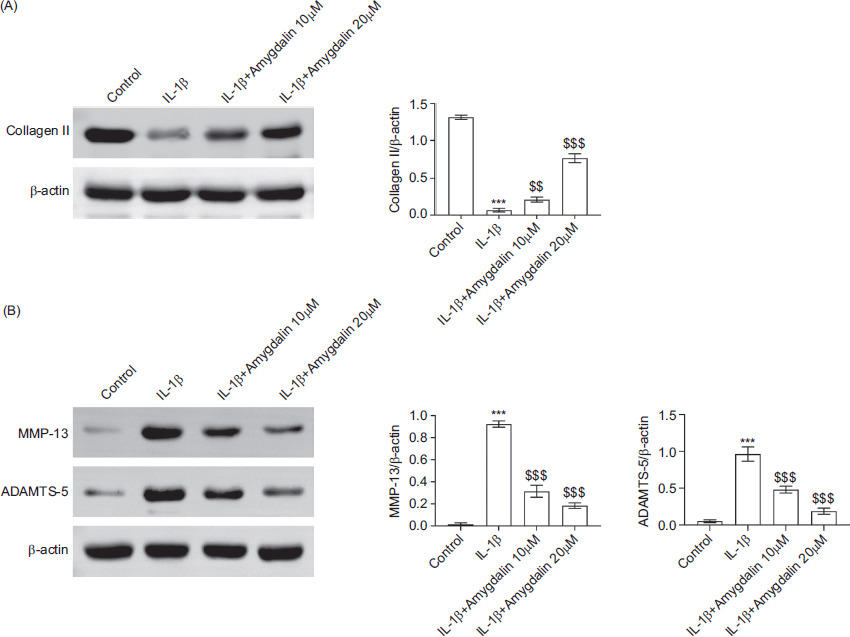
The expression levels of key matrix-associated proteins were examined to evaluate ECM degradation. The results indicated that IL-1β stimulation resulted in a significant reduction in collagen II expression, which was restored upon amygdalin treatment at 10 μM or 20 μM (Figure 3A). Moreover, the expression levels of matrix-degrading enzymes MMP-13 and ADAMTS-5 were markedly increased in response to IL-1β; these increases were significantly mitigated by amygdalin (Figure 3B). These results indicate that amygdalin attenuates IL-1β-mediated ECM degradation in chondrocytes.
Figure 3 Amygdalin inhibits IL-1β-mediated extracellular matrix degradation in chondrocytes. Chondrocytes were divided into control, IL-1β, IL-1β+amygdalin (10 μM), and IL-1β+amygdalin (20 μM) groups. (A) Protein expression of collagen II was detected by Western blot analysis. (B) Protein expression levels of MMP-13 and ADAMTS-5 were analyzed by Western blot analysis. N = 3 repetitions. ***P < 0.001 vs the control group; $$P < 0.01, $$$P < 0.001 vs the IL-1β group.
Amygdalin modulated Nrf2 and NF-κB pathways
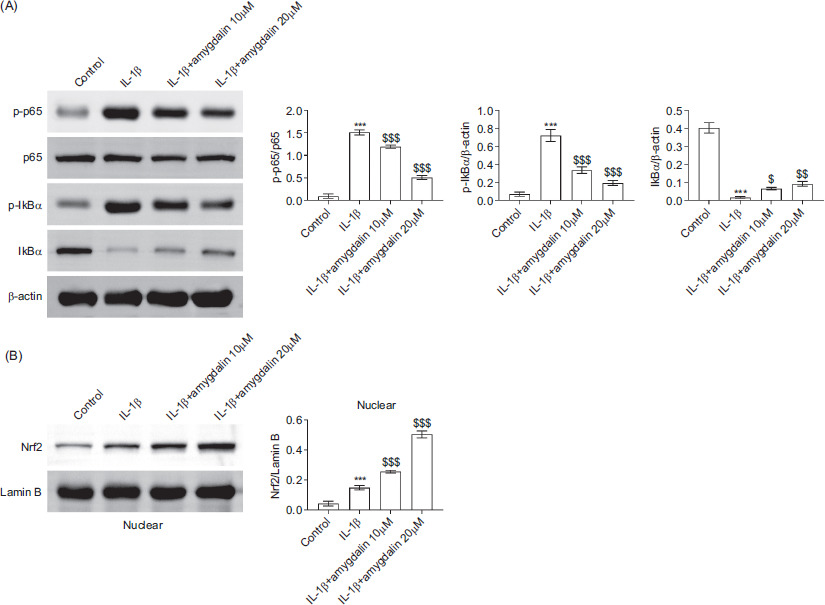
Stimulation of IL-1β enhanced the phosphorylation of p65 protein and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα) and reduced total IκBα levels, indicative of NF-κB activation. Amygdalin treatment at 10 μM or 20 μM reversed these changes, suggesting the inhibition of NF-κB pathway (Figure 4A). In parallel, Nrf2 protein expression was modestly increased following IL-1β stimulation and was further elevated by amygdalin treatment (Figure 4B). Together, these findings indicated that amygdalin exerts its protective effects by activating Nrf2 pathway and suppressing NF-κB signaling in IL-1β-stimulated chondrocytes.
Figure 4 Amygdalin modulates the Nrf2 and NF-κB signaling pathways in IL-1β-stimulated chondrocytes. Chondrocytes were divided into control, IL-1β, IL-1β+amygdalin (10 μM), and IL-1β+amygdalin (20 μM) groups. (A) Protein levels of p-p65, total p65, p-IκBα, and total IκBα were detected by Western blot analysis. (B) Nuclear expression of Nrf2 was measured by Western blot analysis. N = 3 repetitions. ***P < 0.001 vs the control group; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs the IL-1β group.
Discussion
Amygdalin has been reported to possess anti-inflammatory, antioxidant, and immunomodulatory properties.6 However, its regulatory role in the progression of OA has remained insufficiently defined. Amygdalin is a naturally occurring cyanogenic compound, and it’s metabolism in the body can release hydrogen cyanide (HCN), which has certain potential toxicity. Therefore, in this study, when using amygdalin, its toxicological characteristics were evaluated carefully, and cell viability was significantly reduced following amygdalin treatment at concentrations of 40 μM or 80 μM for 24 h and 48 h, indicating cytotoxicity at these doses so that 10 μM or 20 μM of amygdalin was selected for subsequent experiments.
Inflammation plays a critical role in OA pathogenesis, and considerable research has focused on targeting inflammatory pathways to mitigate progression of disease. For instance, achyranthoside D is shown to alleviate inflammation and prevent chondrocyte loss in OA by targeting Wnt3a.12 Similarly, hederagenin attenuates inflammatory response and cartilage degradation, thereby delaying development of OA.13 Labisia pumila has demonstrated the ability to reduce joint inflammation and inhibit collagen breakdown in OA,14 while casticin suppresses the nucleotide-binding domain, leucine rich-containing family, pyrin domain containing 3–hypoxia-inducible factor 1 alpha (HIF-1α/NLRP3) inflammasome signaling axis to prevent monoiodoacetate-induced knee OA.15 In line with these findings, the present study demonstrated that amygdalin effectively attenuates IL-1β-induced inflammation in chondrocytes.
Oxidative stress, characterized by the excessive accumulation of ROS, contributes significantly to OA pathophysiology by promoting cellular damage and cartilage degeneration.16 Concurrently, degradation of ECM is recognized as a hallmark of OA progression.17 Several compounds have been shown to mitigate oxidative stress and prevent ECM breakdown. Emodin, for example, reduces both oxidative stress and ECM degradation in OA,18 while bardoxolonemethyl is shown to protect cartilage by inhibiting oxidative stress-induced ECM damage.19 Additionally, fibroblast growth factor 21 (FGF21) alleviates OA symptoms by suppressing oxidative stress and preserving ECM integrity,20 and methyl gallate counteracts oxidative stress-induced ECM degradation by restoring autophagic activity.21 Consistent with these reports, our findings indicated that amygdalin suppresses IL-1β-induced oxidative stress and ECM degradation in chondrocytes.
Elevated ROS levels are implicated in the activation of multiple signaling pathways that drive inflammation, metabolic dysregulation, and apoptosis, all of which are associated with progression of OA.22 Nrf2 is a pivotal transcription factor that regulates the expression of antioxidant genes.23 Under oxidative stress conditions, Nrf2 translocates into the nucleus and binds to antioxidant response elements (ARE), initiating the transcription of genes involved in cellular defense mechanisms against oxidative damage, inflammation, and apoptosis.24 Notably, Nrf2 deficiency is associated with exacerbated cartilage destruction,25 whereas Nrf2 activation is reported to inhibit IL-1β-induced NF-κB activation in chondrocytes.26 In the present study, we observed that amygdalin enhanced Nrf2 pathway activation while suppressing NF-κB signaling, suggesting that its protective effects in OA could be mediated through Nrf2/NF-κB axis.
Conclusion
This study is the first to reveal that amygdalin attenuates IL-1β-induced chondrocyte damage by modulating the Nrf2/NF-κB signaling pathway, thereby ameliorating OA progression. However, several limitations should be acknowledged. First, the current study lacks validation in clinical specimens and in vivo OA models. Second, the effects of amygdalin on different OA phenotypes and its long-term safety remain to be elucidated fully. The nontoxic concentrations selected based on previous cytotoxicity experiments showed no significant toxicity to cells at concentrations of 10- and 20-μM amygdalin under in vitro conditions, making them suitable for subsequent experiments. The future studies must evaluate the toxicological properties of amygdalin in animal models, including acute toxicity, chronic toxicity, pharmacokinetics, and safety pharmacology, to determine its safe range of dose in the treatment of OA and provide scientific basis for clinical application. The current work revealed the protective effect and potential mechanism of amygdalin on IL-1β-evoked chondrocyte injury in mice through in vitro experiments, but was not confirmed in animal models. The future investigations must focus on addressing these limitations through additional experiments involving OA mice models and clinical samples to further define the therapeutic potential of amygdalin in treating OA.
Ethics Approval
Ethical approval was obtained from the Ethics Committee of Wuhan Hankou Hospital, China.
Data Availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Author Contributions
Li Zhang and Yan Li designed the study and conducted the same. They supervised data collection, and analyzed and interpreted the data. Both authors prepared the manuscript for publication and reviewed the draft of the manuscript. Both authors had read and approved the final manuscript.
Conflcits of Interest
The authors stated that there was no conflict of interest to disclose.
Funding
None.
REFERENCES
1 Abramoff B, Caldera FE. Osteoarthritis: Pathology, diagnosis, and treatment options. Med Clin North Am. 2020;104(2):293–311. 10.1016/j.mcna.2019.10.007
2 Courties A, Kouki I, Soliman N, Mathieu S, Sellam J. Osteoarthritis year in review 2024: Epidemiology and therapy. Osteoarthritis Cartilage. 2024;32(11):1397–404. 10.1016/j.joca.2024.07.014
3 Yao Q, Wu X, Tao C, Gong W, Chen M, Qu M, et al. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct Target Ther. 2023;8(1):56. 10.1038/s41392-023-01330-w
4 Yunus MHM, Nordin A, Kamal H. Pathophysiological perspective of osteoarthritis. Medicina (Kaunas). 2020;56(11):614. 10.3390/medicina56110614
5 Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: A review. JAMA. 2021;325(6):568–78. 10.1001/jama.2020.22171
6 He XY, Wu LJ, Wang WX, Xie PJ, Chen YH, Wang F. Amygdalin–A pharmacological and toxicological review. J Ethnopharmacol. 2020;254:112717. 10.1016/j.jep.2020.112717
7 Wang Z, Du H, Wan H, Yang J, Wan H. Amygdalin prevents multidrug-resistant Staphylococcus aureus-induced lung epithelial cell injury by regulating inflammation and oxidative stress. PLoS One. 2024;19(9):e0310253. 10.1371/journal.pone.0310253
8 Zeng Q, Sun Q, Xu H, Chen J, Ling H, Ge Q, et al. Amygdalin delays cartilage endplate degeneration and improves intervertebral disc degeneration by inhibiting NF-κB signaling pathway and inflammatory response. J Inflamm Res. 2023;16:3455–68. 10.2147/JIR.S415527
9 He X, Liu J, Gong Y, Lu W, Sha X, Cao C, et al. Amygdalin ameliorates alopecia areata on C3H/HeJ mice by inhibiting inflammation through JAK2/STAT3 pathway. J Ethnopharmacol. 2024;331:118317. 10.1016/j.jep.2024.118317
10 Li S, Lu S, Wang L, Liu S, Zhang L, Du J, et al. Effects of amygdalin on ferroptosis and oxidative stress in diabetic retinopathy progression via the NRF2/ARE signaling pathway. Exp Eye Res. 2023;234:109569. 10.1016/j.exer.2023.109569
11 Trang NM, Kim EN, Lee HS, Jeong GS. Effect on osteoclast differentiation and ER stress downregulation by amygdalin and RANKL binding interaction. Biomolecules. 2022;12(2):256. 10.3390/biom12020256
12 Xie W, Qi S, Dou L, Wang L, Wang X, Bi R, et al. Achyranthoside D attenuates chondrocyte loss and inflammation in osteoarthritis via targeted regulation of Wnt3a. Phytomedicine. 2023;111:154663. 10.1016/j.phymed.2023.154663
13 Shen Y, Teng L, Qu Y, Huang Y, Peng Y, Tang M, et al. Hederagenin suppresses inflammation and cartilage degradation to ameliorate the progression of osteoarthritis: An in vivo and in vitro study. Inflammation. 2023;46(2):655–78. 10.1007/s10753-022-01763-5
14 Madzuki IN, Lau SF, Che Ahmad Tantowi NA, Mohd Ishak NI, Mohamed S. Labisia pumila prevented osteoarthritis cartilage degeneration by attenuating joint inflammation and collagen breakdown in postmenopausal rat model. Inflammopharmacology. 2018;26(5):1207–17. 10.1007/s10787-018-0452-6
15 Li X, Mei W, Huang Z, Zhang L, Zhang L, Xu B, et al. Casticin suppresses monoiodoacetic acid-induced knee osteoarthritis through inhibiting HIF-1α/NLRP3 inflammasome signaling. Int Immunopharmacol. 2020;86:106745. 10.1016/j.intimp.2020.106745
16 Zhang S, Wang L, Kang Y, Wu J, Zhang Z. Nanomaterial-based reactive oxygen species scavengers for osteoarthritis therapy. Acta Biomater. 2023;162:1–19. 10.1016/j.actbio.2023.06.012
17 Rapp AE, Zaucke F. Cartilage extracellular matrix-derived matrikines in osteoarthritis. Am J Physiol Cell Physiol. 2023;324(2):C377–94. 10.1152/ajpcell.00464.2022
18 Ma T, Ma Y, Yu Y, Jia L, Lv L, Song X, et al. Emodin attenuates the ECM degradation and oxidative stress of chondrocytes through the Nrf2/NQO1/HO-1 pathway to ameliorate rat osteoarthritis. Oxid Med Cell Longev. 2022;2022:5581346. 10.1155/2022/5581346
19 Pang Z, Jiang Z, Zhu R, Song C, Tang H, Cao L, et al. Bardoxolone-methyl prevents oxidative stress-mediated apoptosis and extracellular matrix degradation in vitro and alleviates osteoarthritis in vivo. Drug Des Devel Ther. 2021;15:3735–47. 10.2147/DDDT.S314767
20 Lu H, Jia C, Wu D, Jin H, Lin Z, Pan J, et al. Fibroblast growth factor 21 (FGF21) alleviates senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the SIRT1-mTOR signaling pathway. Cell Death Dis. 2021;12(10):865. 10.1038/s41419-021-04157-x
21 Li Y, Shen B, Lv C, Zhu X, Naren Q, Xu D, et al. Methyl gallate prevents oxidative stress induced apoptosis and ECM degradation in chondrocytes via restoring Sirt3 mediated autophagy and ameliorates osteoarthritis progression. Int Immunopharmacol. 2023;114:109489. 10.1016/j.intimp.2022.109489
22 Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862(4):576–91. 10.1016/j.bbadis.2016.01.003
23 He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21(13):4777. 10.3390/ijms21134777
24 Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73(17):3221–47. 10.1007/s00018-016-2223-0
25 Ferrándiz ML, Nacher-Juan J, Alcaraz MJ. Nrf2 as a therapeutic target for rheumatic diseases. Biochem Pharmacol. 2018;152:338–46. 10.1016/j.bcp.2018.04.010
26 Zhang Z, Wang S, Liu X, Yang Y, Zhang Y, Li B, et al. Secoisolariciresinol diglucoside ameliorates osteoarthritis via nuclear factor-erythroid 2-related factor-2/nuclear factor kappa B pathway: In vitro and in vivo experiments. Biomed Pharmacother. 2023;164:114964. 10.1016/j.biopha.2023.114964