
Download
ORIGINAL ARTICLE
Knockdown of ARHGDIB promotes autophagy and reduces inflammation in LPS-induced alveolar epithelial cells via the PRKACB/NF-κB pathway
Haizhen Jin, Shuangxia Dong*, Zhihui Li, Xinjian Dai
Department of Respiratory and Critical Care Medicine, the Wenzhou Central Hospital and Dingli Clinical Institute of Wenzhou Medical University, Wenzhou, Zhejiang, China
Abstract
Background: Acute lung injury (ALI) is a critical clinical condition with high mortality, necessitating the development of more effective therapeutic strategies. Rho Guanine nucleotide dissociation inhibitor (GDP) beta (ARHGDIB) has been shown to exert protective effects against noxious stimuli in various disease models.
Objective: In this study, we investigated whether ARHGDIB knockdown had a protective effect on lipopolysaccharide (LPS)-induced injury in alveolar epithelial cells and elucidated its underlying molecular mechanisms.
Material and Methods: Mouse alveolar epithelial cells that were isolated from the lung of a 5-month-old female mouse (MLE-12) were treated with LPS, followed by ARHGDIB knockdown and overexpression of protein kinase A (PKA)-activated catalytic subunit β (PRKACB). Oxidative stress and apoptosis were assessed, while inflammatory cytokine levels were quantified using enzyme-linked immunosorbent serologic assays. Autophagy and PRKACB/nuclear factor kappa B (NF-κB) pathway activation was evaluated by Western blot analysis. Results: LPS upregulated ARHGDIB expression in alveolar epithelial cells. Silencing ARHGDIB significantly reduced oxidative stress inflammation, and promoted autophagy in LPS-treated MLE-12 cells. ARHGDIB knockdown modulated the PRKACB/NF-κB signaling pathway, thereby promoting autophagy and alleviating LPS-induced cellular injury.
Conclusion: This regulatory mechanism significantly reduced oxidative stress and inflammatory responses in alveolar epithelial cells, highlighting the protective role of ARHGDIB silencing in LPS-induced lung injury.
Key words: ARHGDIB, inflammation oxidative stress, PKA, PRKACB/NF-κB acute lung injury autophagy
*Corresponding author: Shuangxia Dong, Department of Respiratory and Critical Care Medicine, the Wenzhou Central Hospital and Dingli Clinical Institute of Wenzhou Medical University, No. 252, Baili East Road, Lucheng District, Wenzhou 325000, Zhejiang Province, China. Email address: [email protected]
Received 14 March 2025; Accepted 27 May 2025; Available online 1 September 2025
Copyright: Jin H, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Acute lung injury (ALI) is a severe and debilitating condition characterized by widespread pulmonary inflammation and impaired oxygen uptake in the lungs.1 The progression of ALI involves a pronounced inflammatory response that leads to cellular apoptosis, necrosis, and release of fibrotic mediators.2 The endotoxin lipopolysaccharide (LPS) is produced from the outer membrane of Gram-negative bacteria. It induces ALI through toll-like receptor 4 (TLR4)-mediated oxidative stress and inflammation, and is widely used as a standard agent to induce ALI in experimental models.3 To date, existing therapies for ALI are often limited in their efficacy and may cause significant adverse effects. Consequently, there is an urgent need to develop novel treatment strategies that are both effective and associated with minimal adverse effects.
Autophagy is a critical intracellular degradation mechanism responsible for the clearance of damaged proteins and organelles. It plays a vital role in regulating lung injury and tissue repair processes.4 Emerging studies suggest that inhibiting autophagy can exacerbate LPS-induced ALI in human bronchial epithelial cells via nuclear factor kappa B (NF-κB) signaling pathway.5 Conversely, rapamycin (RAPA), a well-known autophagy activator, has been shown to mitigate LPS-induced lung injury and suppress the associated inflammatory response.6 Based on this evidence, targeting autophagy represents a promising therapeutic strategy for the treatment of ALI.
A recent study utilized a consensus machine-learning approach to diagnose sepsis-induced ALI and identified Rho Guanine nucleotide dissociation inhibitor (GDP) beta (ARHGDIB) as a potential biomarker for ALI.7 ARHGDIB is a pivotal molecule in cell signaling, predominantly expressed in hematopoietic tissues, including B and T lymphocyte cell lines.8 Past studies have shown that by modifying the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in phagocytic cells, overexpression of ARHGDIB can improve macrophage infiltration and increase the generation of reactive oxygen species (ROS).9 Moreover, ARHGDIB has been shown to inhibit endothelial cell migration and modulate various vascular functions, including the regulation of vascular tone.10 These findings suggest that ARHGDIB may contribute to the pathogenesis and progression of ALI through its effects on immune responses and vascular homeostasis.
In light of these findings, the role and mechanisms of ARHGDIB signaling in the context of ALI remain unclear. Thus, in this work, we assessed the impact of ARHGDIB knockdown on LPS-induced autophagy, inflammation, and oxidative stress in alveolar epithelial cells.
Methods
Cell culture
Mouse alveolar epithelial cells (MLE-12) were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal calf serum and maintained at 37°C in a humidified incubator containing 5% CO2. For LPS stimulation, cells were treated with 1-µg/mL LPS (Sigma, MA, USA) and incubated for 12 h.
Cell transfection
Small interfering RNA (siRNA)-mediated gene silencing was performed using either negative control siRNA (si-NC, 5′-GTCGAACGTCGTGAACCTACCATG-3′) or ARHGDIB-specific siRNA (si-ARHGDIB, 5′-GGAAGGTTCTGAATATAGA-3′) transfected into cells with lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s protocol. Overexpression of protein kinase A (PKA)-activated catalytic subunit β (PRKACB) was achieved by transfecting cells with plasmid cloning DNA (pcDNA)–PRKACB plasmid using the same reagent.
Apoptosis analysis
In 24-well plates, cells were seeded at a density of 2×105 cells/well (n = 3), followed by LPS stimulation and transfection as indicated. Apoptosis was assessed using the Annexin V-Enhanced Green Fluorescent Protein (EGFP) apoptosis detection kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol. Samples were analyzed with a BD Accuri™ C6 Plus flow cytometer.
Enzyme-linked immunosorbent serologic assay (ELISA)
Cells were seeded in 12-well plates at a density of 2×105 cells/well (n = 3). After stimulation with LPS and transfection, culture supernatants were collected, and cytokine levels of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) were quantified using ELISA kits (Beyotime) according to the manufacturer’s protocol.
Detecting oxidative stress damage
Reactive oxygen species (ROS) were measured with a dihydroethidium fluorescent probe (KeyGen Biotech, Nanjing, China). Following the treatment, cells were harvested and analyzed for oxidative stress markers. Superoxide dismutase (SOD) activity and malondialdehyde (MDA) levels were determined using commercial assay kits (Beyotime) according to the manufacturer’s protocol.
Immunofluorescence
After fixing the cells with 4% paraformaldehyde, they were permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Following this, the cells were blocked with 10% bovine serum for 30 min to prevent nonspecific binding. Subsequently, the cells were incubated with antibodies targeting ARHGDIB (ab181252; Abcam, Cambridge, UK) and LC3B (ab192890; Abcam), both diluted in 1:200 ratio overnight at 4°C. After three washes with phosphate-buffered saline solution (PBS), the cells were treated for an hour at room temperature with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min, and fluorescence signals were visualized using a fluorescence microscope.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Real-time quantitative polymerase chain reaction assays were performed according to previous studies.11 The sequences used were as follows: ARHGDIB, “F” variant (ARHGDIB-F) 5′-ctcggcctgaggagtatgag-3′; ARHGDIB, “R” variant (ARHGDIB-R) 5′-gtggtcttgcttgtcatcgt-3′; PRKACB “F” variant (PRKACB-F) 5′-AGTGGTTTGCCACGACAGATTG-3′; PRKACB “R” variant (PRKACB-R) 5′-TTGCTGGTACCAGAGCCTCTAA-3′; glyceraldehyde 3-phosphate dehydrogenase, “F” variant (GAPDH-F) 5′-GCACCGTCAAGGCTGAGAAC-3′; GAPDH, “R” variant (GAPDH-R), 5′-TGGTGAAGACGCCAGTGGA-3′.
Western blot analysis
A bicinchoninic acid (BCA) protein assay kit (Beyotime) was used to determine protein concentration after cells were lysed in radioimmunoprecipitation assay (RIPA) buffer. Equal amount of protein was separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. The membrane was blocked with 5% non-fat dry milk and then incubated with the following listed primary antibodies: ARHGDIB (ab181252; Abcam), LC3B (ab192890; Abcam), Beclin-1 (ab302669; Abcam), p62 (ab109012; Abcam), PRKACB (ab258603; Abcam), p65 (ab32536; Abcam), p-p65 (ab53489; Abcam), Ikb-α (ab32518; Abcam), p-Ikb-α (ab92700; Abcam), and GAPDH (ab9485; Abcam), and incubated overnight at 4°C. Secondary antibodies labeled with peroxidase were added to the membrane after it was washed. Using an enhanced chemiluminescence (ECL) detection kit and quantified using the ImageJ software, GAPDH was used for normalization.
Immunofluorescent staining for p65
Cells were fixed with 4% paraformaldehyde and incubated with rabbit monoclonal antibody against p65 (cat. #4764; Cell Signaling Technology [CST], Beverly, MA, USA). After washing, cells were incubated with a HiLyte Fluor 555-conjugated goat anti-rabbit immunoglobulin G (IgG) (AnaSpec, Fremont, CA, USA). Nuclei were counterstained with DAPI. A Zeiss Axio Imager Z1 (Zeiss, Jena, Germany) was used to capture fluorescence images.
Statistical analysis
Data were analyzed using the SPSS software, version 19.0 (SPSS Inc., Chicago, IL, USA). Results were presented as the mean ± standard deviation (SD). Two-tailed Student’s t-test was used for comparisons between two groups, while one-way ANOVA was used for comparisons between multiple groups. Post hoc testing was performed using Tukey’s multiple comparison test; P < 0.05 was considered statistically significant. All experiments were independently repeated for three times.
Results
LPS induced high expression of ARHGDIB in alveolar epithelial cells
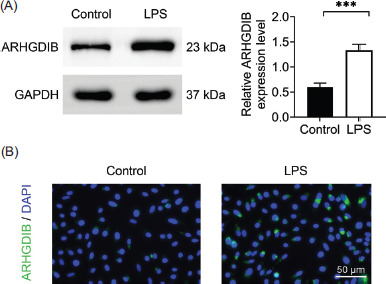
In order to investigate the effect of LPS on ARHGDIB expression, mouse alveolar epithelial MLE-12 cells were treated with LPS for 12 h. Western blot analysis revealed a significant upregulation of ARHGDIB protein levels in response to LPS stimulation (Figure 1A). Consistent with these findings, immunofluorescence staining demonstrated increased ARHGDIB fluorescence intensity in the LPS-treated group, compared to controls (Figure 1B), confirming the elevated expression of ARHGDIB under inflammatory conditions.
Figure 1 LPS-induced high expression of ARHGDIB in alveolar epithelial cells. (A) Detection of ARHGDIB expression in alveolar epithelial cells by Western blot analysis. (B) Immunofluorescence examination of ARHGDIB fluorescence expression in alveolar epithelial cells. Values are presented as mean ± SD. ***P < 0.001; n = 3.
Knockdown of ARHGDIB alleviated LPS-induced oxidative stress and inflammatory damage in alveolar epithelial cells
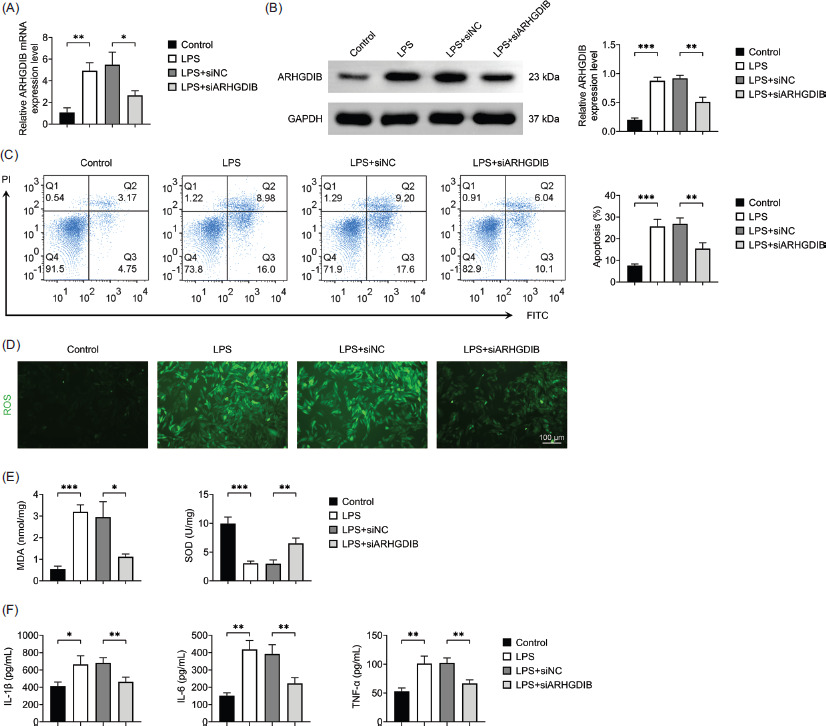
In order to confirm successful silencing of ARHGDIB, MLE-12 cells were transfected with ARHGDIB-specific siRNA, and protein expression was assessed by Western blot analysis. A marked reduction in ARHGDIB expression was observed in transfected cells, indicating effective knockdown (Figures 2A and 2B). Increased oxidative stress is a key driver in the pathogenesis of ALI, marked by an imbalance between oxidants, including ROS and MDA, and antioxidants, such as SOD. In response to LPS stimulation, MLE-12 cells exhibited significantly increased apoptosis, ROS production, and MDA levels along with reduced SOD activity. Notably, ARHGDIB knockdown reversed these changes, significantly mitigating oxidative damage (Figures 2C–2E).
Figure 2 Knockdown of ARHGDIB-alleviated LPS-induced oxidative stress and inflammatory damage in alveolar epithelial cells. (A) The mRNA expression of ARHGDIB in alveolar epithelial cells was detected by RT-qPCR. (B) Detection of ARHGDIB expression in alveolar epithelial cells by Western blot analysis. Detection of ARHGDIB expression in alveolar epithelial cells by Western blot analysis. (C) Detection of apoptosis rate by flow cytometry. (D) Dihydroethidium fluorescent probe to check ROS fluorescence expression. (E) Kit detects MDA and SOD levels in alveolar epithelial cells. (F) ELISA detects IL-1β, IL-6, and TNF-α levels in alveolar epithelial cells culture fluid. Values are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001; n = 3.
Excessive infiltration of inflammatory cells and dysregulated cytokine secretion are intricately linked to the pathogenesis of ALI. Our results demonstrated that LPS treatment markedly elevated the secretion of IL-1β, IL-6, and TNF-α, while ARHGDIB knockdown significantly suppressed the levels of these cytokines (Figure 2F). These results indicate that knockdown of ARHGDIB can alleviate LPS-induced oxidative stress and inflammatory injury in alveolar epithelial cells.
Knockdown of ARHGDIB enhanced autophagy inhibited by LPS in alveolar epithelial cells
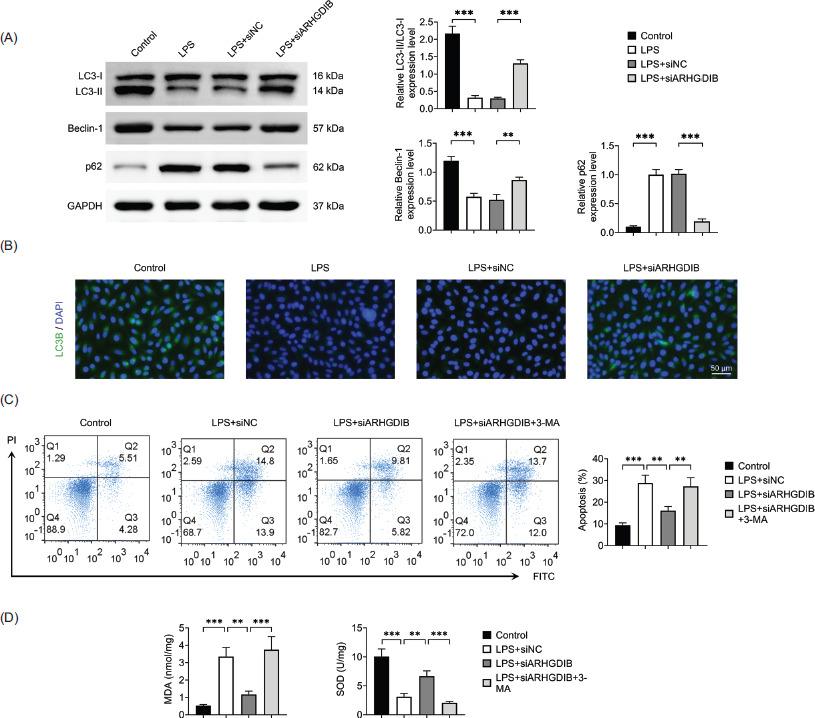
Autophagy plays a complex role in lung diseases, exerting both protective and detrimental effects depending on the context.12 To determine whether ARHGDIB regulates autophagy in LPS-induced injury, we assessed autophagic flux in MLE-12 cells. LC3 is a well-established marker of autophagy, where the conversion of LC3-I to LC3-II reflects autophagosome formation. Western blot analysis revealed that LPS stimulation significantly increased the expression of p62, a substrate of autophagic degradation while decreasing Beclin-1 expression and the LC3-II:LC3-I ratio. However, knocking down of ARHGDIB reversed these effects of LPS (Figure 3A). Immunofluorescence analysis was further employed to detect autophagy activation. The results revealed that LC3 punctate formation was increased following ARHGDIB knockdown (Figure 3B). To investigate the functional relevance of autophagy in the protective effects of ARHGDIB silencing, the autophagy inhibitor 3-methyladenine (3-MA, 5 mM) was applied.13 Treatment with 3-MA significantly reversed the reduction in apoptosis and oxidative stress observed following ARHGDIB knockdown (Figures 3C and 3D), indicating that the protective effects of ARHGDIB silencing are mediated through autophagy activation.
Figure 3 ARHGDIB enhanced LPS-induced autophagy inhibited by LPS in alveolar epithelial cells. (A) Western blot analysis to detect the expression of LC3II/I, p62, and Beclin-1 in alveolar epithelial cells. (B) Immunofluorescence examination of LC3B fluorescence expression in alveolar epithelial cells. (C) Detection of apoptosis rate by flow cytometry. (D) Kit detects MDA and SOD levels in alveolar epithelial cells. Values are presented as mean ± SD. **P < 0.01, ***P < 0.001; n = 3.
ARHGDIB modulated the PRKACB/NF-κB pathway
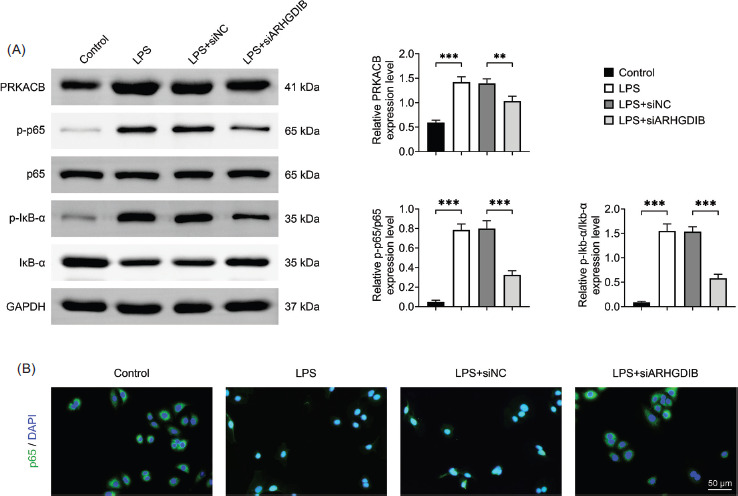
Next, we explore the molecular mechanism by which ARHGDIB exerts its effects in regulating the PRKACB/NF-κB signaling pathway. The findings demonstrated that alveolar epithelial cells’ expression of PRKACB and phosphorylation of p65 and Ikb-α increased in LPS environment whereas the same cells’ expression and phosphorylation of p65 and Ikb-α decreased following the knockdown of ARHGDIB (Figure 4A). We consistently observed significant nuclear translocation of NF-κB in LPS-treated cells. However, knockdown of ARHGDIB effectively blocked this translocation, further supporting its role in modulating NF-κB signaling (Figure 4B).
Figure 4 ARHGDIB modulated the PRKACB/NF-κB pathway. (A) Western blot analysis to detect PRKACB, p65, p-p65, Ikb-α, and p-Ikb-α protein expression. (B) Immunofluorescence staining image of p65. Values are presented as mean ± SD. **P < 0.01, ***P < 0.001; n = 3.
PRKACB overexpression reversed the protective effects of ARHGDIB silencing in LPS-stimulated alveolar epithelial cells
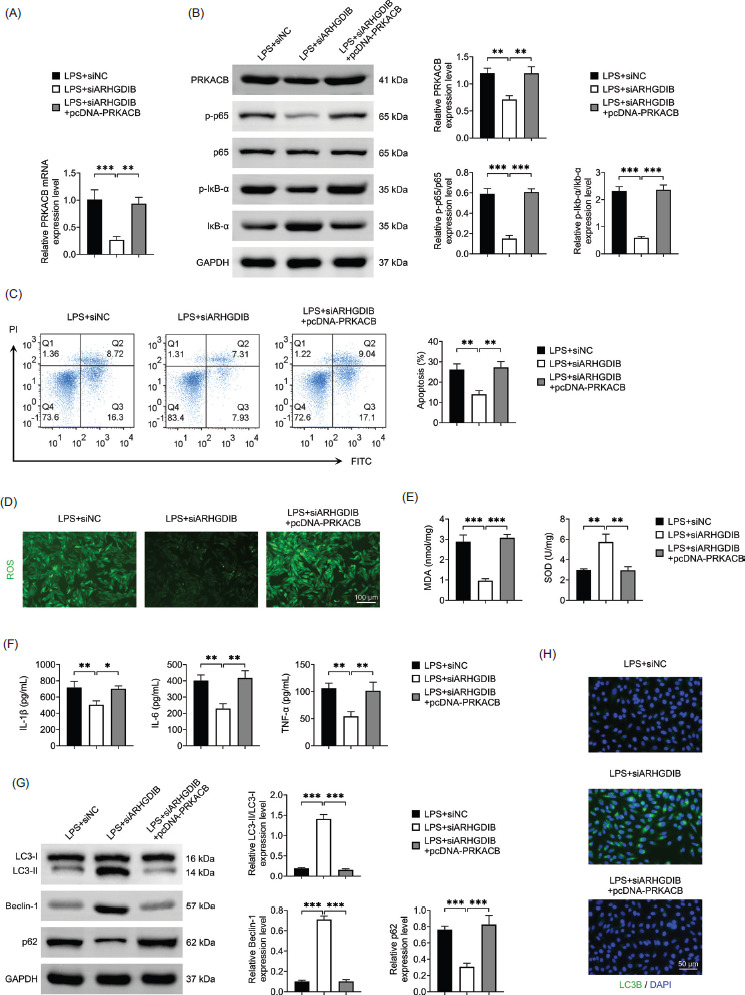
In order to explore whether the role of ARHGDIB is related to PRKACB, we overexpressed PRKACB. As confirmed by Western blot analysis, PRKACB was successfully overexpressed in alveolar epithelial cells, which led to reactivation of the NF-κB signaling pathway (Figures 5A and 5B).
Functionally, PRKACB overexpression reversed the antioxidant and anti-inflammatory effects induced by ARHGDIB knockdown. Specifically, LPS-induced oxidative stress and inflammatory cytokine production were reinstated upon PRKACB overexpression in cells with ARHGDIB knockdown (Figures 5C–5F). Moreover, the autophagy-promoting effect of ARHGDIB silencing was also attenuated by PRKACB overexpression, as indicated by reduced LC3-II levels and increased p62 expression (Figures 5G and 5H). These findings suggest that knocking down of ARHGDIB promotes autophagy by regulating the PRKACB/NF-κB pathway, thereby improving the oxidative stress and inflammatory response of LPS-induced alveolar epithelial cells.
Figure 5 PRKACB overexpression reversed the protective effects of ARHGDIB silencing in LPS-stimulated alveolar epithelial cells. (A) The mRNA expression of PRKACB in alveolar epithelial cells was detected by RT-qPCR. (B) Western blot analysis to detect PRKACB, p65, p-p65, Ikb-α, and p-Ikb-α protein expression. (C) Detection of apoptosis rate by flow cytometry. (D) Dihydroethidium fluorescent probe to check ROS fluorescence expression. (E) Kit detects MDA and SOD levels in alveolar epithelial cells. (F) ELISA detects IL-1β, IL-6, and TNF-α levels in alveolar epithelial cells culture fluid. (G) Western blot analysis to detect the expression of LC3II/I, p62, and Beclin-1 in alveolar epithelial cells. (H) Immunofluorescence examination of LC3B fluorescence expression in alveolar epithelial cells. Values are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001; n = 3.
Discussion
Acute lung injury is a severe pulmonary condition associated with high mortality, often triggered by infection, trauma, and chemical exposure.14 When the lungs are exposed to endogenous or exogenous insults, alveolar macrophages are activated initially. These cells release pro-inflammatory mediators, which in turn stimulate alveolar macrophages, epithelial cells, and fibroblasts to produce excessive ROS.15 Recent studies have identified ARHGDIB as a contributor to the pathogenesis of ALI.7,16 While ARHGDIB has been shown to exert regulatory effects on alveolar macrophages, its role in alveolar epithelial cells remains unclear.17 In this study, LPS caused high expression of ARHGDIB in alveolar epithelial cells, and knocking down ARHGDIB promoted autophagy, thereby improving inflammation and oxidative stress damage caused by LPS.
An abnormal inflammatory response is a key factor in the development and progression of ALI, especially in LPS-induced cases. The balance of pro- and anti-inflammatory cytokines in the ALI microenvironment is a key determinant of lung tissue injury and repair.18 Among these cytokines, TNF-α is known to accelerate the migration of neutrophils to the sites of injury, enhance the activity of proteolytic enzyme , and elevate the production of reactive ROS, all of which contribute to the lung injury.19 In addition, IL-1β and IL-6, two potent pro-inflammatory mediators, further activate immune cells and promote the release of downstream inflammatory factors.20 Alveolar epithelial cells themselves contribute to this process by generating excessive ROS during respiratory bursts.15 The continuous production of ROS leads to severe oxidative stress, which is a critical factor in accelerating the onset and progression of ALI.21 Our study showed that the levels of inflammatory factors and oxidative stress in alveolar epithelial cells were significantly reduced after inhibiting ARHGDIB in the LPS environment. This discovery implies that oxidative stress brought on by exposure to LPS and the release of inflammatory mediators may be mediated by ARHGDIB.
The role of autophagy in lung diseases remains a subject of ongoing debate; however, growing evidence suggests that mild activation of autophagy may be beneficial in certain pathological contexts. For example, rapamycin, a well-known autophagy inducer, significantly reduces oxidative stress and lung inflammation in ALI mice by enhancing autophagic flux. In contrast, pharmacological inhibition of autophagy has been reported to aggravate LPS-induced lung injury.22,23 In addition, studies have shown that ARHGDIB is a therapeutic target for autophagy in lupus nephritis.24 In our study, we observed that autophagy was suppressed in alveolar epithelial cells upon LPS exposure, as evidenced by decreased LC3-II levels and increased p62 expression. However, knockdown of ARHGDIB restored autophagic activity, suggesting that ARHGDIB may contribute to LPS-induced autophagy inhibition in alveolar epithelial cells. The PRKACB gene encodes the catalytic subunit of PKA, a key enzyme involved in intracellular signaling pathways.25 PKA is essential for transducing signals that regulate a wide range of physiological cell functions. Upon exposure to various extracellular stimuli, PKA can activate the NF-κB pathway, thereby participating in inflammatory responses and contributing to cell damage.26 The transcription of pro-inflammatory cytokines is driven by aberrant activation of NF-κB, which intensifies inflammatory response and aids in the development of ALI and other inflammatory illnesses.27 In addition, PKA can also phosphorylate ARHGDIB, which will lead to the inactivation of Rho GTPase, thus blocking the high permeability of endothelial cells.28 Additionally, PKA/NF-κB is crucial for acute lung damage.29 Our findings suggests that ARHGDIB inhibits autophagy via the PRKACB/NF-κB signaling pathway, which, in turn, contributes to LPS-induced inflammation and oxidative stress in alveolar epithelial cells.
This study also has some limitations. The experiments were limited to in vitro investigations using the MLE-12 alveolar epithelial cell line. While this cell line is widely used for mechanistic studies, it does not fully replicate the complexity of the in vivo pulmonary environment. Future studies involving in vivo models and clinical samples are necessary to confirm the therapeutic potential of targeting ARHGDIB in the treatment of ALI.
Conclusion
In summary, our data indicate that knocking down of ARHGDIB can improve LPS-induced inflammation and oxidative stress damage in alveolar epithelial cells by promoting autophagy through regulating the PRKACB/NF-κB signaling pathway. These results highlight ARHGDIB as a potential therapeutic target, and suggest that ARHGDIB-based interventions can offer a promising strategy for the prevention and treatment of ALI.
Availability of Data and Materials
All data generated or analyzed in this study are included in this published article. The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.
Author Contributions
Haizhen Jin: conceptualization, methodology, validation, supervision, and writing of original draft, review, and editing; Shuangxia Dong: formal analysis, resources, and investigation; Zhihui Li: formal analysis, visualization and data curation; Xinjian Dai: project administration, supervision, and validation. All authors had read and approved the final manuscript.
Conflicts of Interest
The authors stated that there was no conflict of interest to declare.
Funding
Not Applicable.
REFERENCES
1 Mowery NT, Terzian WTH, Nelson AC. Acute lung injury. Curr Probl Surg. 2020;57(5):100777. Epub 2020/06/09. 10.1016/j.cpsurg.2020.100777
2 Long ME, Mallampalli RK, Horowitz JC. Pathogenesis of pneumonia and acute lung injury. Clin Sci (Lond). 2022;136(10):747–69. Epub 2022/05/28. 10.1042/CS20210879
3 Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–92. Epub 1999/04/10. 10.1074/jbc.274.16.10689
4 Liu D, Weng S, Fu C, Guo R, Chen M, Shi B, et al. Autophagy in acute lung injury. Cell Biochem Biophys. 2025;83:1415–1425. Epub 2024/11/13. 10.1007/s12013-024-01604-2
5 Hu Y, Lou J, Mao YY, Lai TW, Liu LY, Zhu C, et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12(12):2286–99. Epub 2016/09/23. 10.1080/15548627.2016.1230584
6 Jia X, Cao B, An Y, Zhang X, Wang C. Rapamycin ameliorates lipopolysaccharide-induced acute lung injury by inhibiting IL-1beta and IL-18 production. Int Immunopharmacol. 2019;67:211–9. Epub 2018/12/18. 10.1016/j.intimp.2018.12.017
7 Zheng Y, Wang J, Ling Z, Zhang J, Zeng Y, Wang K, et al. A diagnostic model for sepsis-induced acute lung injury using a consensus machine learning approach and its therapeutic implications. J Transl Med. 2023;21(1):620. Epub 2023/09/13. 10.1186/s12967-023-04499-4
8 Olofsson B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell Signal. 1999;11(8):545–54. Epub 1999/08/05. 10.1016/s0898-6568(98)00063-1
9 Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest. 2012;122(4):1503–18. Epub 2012/03/13. 10.1172/JCI61392
10 Nagar H, Kim S, Lee I, Choi SJ, Piao S, Jeon BH, et al. CRIF1 deficiency suppresses endothelial cell migration via upregulation of RhoGDI2. PLoS One. 2021;16(8):e0256646. Epub 2021/08/27. 10.1371/journal.pone.0256646
11 Zhao N, Wang R, Zhou L, Zhu Y, Gong J, Zhuang SM. MicroRNA-26b suppresses the NF-kappaB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol Cancer. 2014;13:35. Epub 2014/02/26. 10.1186/1476-4598-13-35
12 Kong X, Lu L, Lin D, Chong L, Wen S, Shi Y, et al. FGF10 ameliorates lipopolysaccharide-induced acute lung injury in mice via the BMP4-autophagy pathway. Front Pharmacol. 2022;13:1019755. Epub 2023/01/10. 10.3389/fphar.2022.1019755
13 Hu Q, Liu H, Wang R, Yao L, Chen S, Wang Y, et al. Capsaicin attenuates LPS-induced acute lung injury by inhibiting inflammation and autophagy through regulation of the TRPV1/AKT pathway. J Inflamm Res. 2024;17:153–70. Epub 2024/01/15. 10.2147/JIR.S441141
14 Cao C, Zhang L, Shen J. Phosgene-induced acute lung injury: Approaches for mechanism-based treatment strategies. Front Immunol. 2022;13:917395. Epub 2022/08/20. 10.3389/fimmu.2022.917395
15 Pan P, Su L, Wang X, Chai W, Liu D, Song L, et al. Vimentin regulation of autophagy activation in lung fibroblasts in response to lipopolysaccharide exposure in vitro. Ann Transl Med. 2021;9(4):304. Epub 2021/03/13. 10.21037/atm-20-5129
16 Yang Y, Tian Z, He L, Meng H, Xie X, Yang Z, et al. RhoGDI beta inhibition via miR-200c/AUF1/SOX2/miR-137 axis contributed to lncRNA MEG3 downregulation-mediated malignant transformation of human bronchial epithelial cells. Mol Carcinog. 2024;63(5):977–90. Epub 2024/02/20. 10.1002/mc.23702
17 Yan C, Wang X, Liu Y, Abdulnour RE, Wu M, Gao H. Protective role of Rho guanosine diphosphate dissociation inhibitor, Ly-GDI, in pulmonary alveolitis. PLoS One. 2015;10(10):e0140804. Epub 2015/10/16. 10.1371/journal.pone.0140804
18 Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722. Epub 2020/08/28. 10.3389/fimmu.2020.01722
19 Hu X, Su J, Chen M, Tu Y, Wu C, Cao X, et al. Macrophage-derived exosomal TNF-alpha promotes pulmonary surfactant protein expression in PM(2.5)-induced acute lung injury. Sci Total Environ. 2023;892:164732. Epub 2023/06/09. 10.1016/j.scitotenv.2023.164732
20 Zhang Y, Cui Y, Feng Y, Jiao F, Jia L. Lentinus edodes polysaccharides alleviate acute lung injury by inhibiting oxidative stress and inflammation. Molecules. 2022;27(21):7328. Epub 2022/11/12. 10.3390/molecules27217328
21 Li-Mei W, Jie T, Shan-He W, Dong-Mei M, Peng-Jiu Y. Anti-inflammatory and anti-oxidative effects of dexpanthenol on lipopolysaccharide induced acute lung injury in mice. Inflammation. 2016;39(5):1757–63. Epub 2016/07/30. 10.1007/s10753-016-0410-7
22 Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125(Pt 2):507–15. Epub 2012/02/04. 10.1242/jcs.094573
23 Lorne E, Zhao X, Zmijewski JW, Liu G, Park YJ, Tsuruta Y, et al. Participation of mammalian target of rapamycin complex 1 in toll-like receptor 2-and 4-induced neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol. 2009;41(2):237–45. Epub 2009/01/10. 10.1165/rcmb.2008-0290OC
24 Zhou XJ, Klionsky DJ, Zhang H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy. 2019;15(5):908–12. Epub 2019/02/14. 10.1080/15548627.2019.1580512
25 Newman T, Bond DM, Ishihara T, Rizzoli P, Gouil Q, Hore TA, et al. PRKACB is a novel imprinted gene in marsupials. Epigenetics Chromatin. 2024;17(1):29. Epub 2024/09/29. 10.1186/s13072-024-00552-8
26 Xu F, Wang J, Cao Z, Song M, Fu Y, Zhu Y, et al. cAMP/PKA signaling pathway induces apoptosis by inhibited NF-kappaB in aluminum chloride-treated lymphocytes in vitro. Biol Trace Elem Res. 2016;170(2):424–31. Epub 2015/08/19. 10.1007/s12011-015-0461-x
27 Zhao W, Wang J, Li X, Li Y, Ye C. Deoxycholic acid inhibits Staphylococcus aureus-induced endometritis through regulating TGR5/PKA/NF-kappaB signaling pathway. Int Immunopharmacol. 2023;118:110004. Epub 2023/03/24. 10.1016/j.intimp.2023.110004
28 Chen H, Shen Y, Liang Y, Qiu Y, Xu M, Li C. Selexipag improves lipopolysaccharide-induced ARDS on C57BL/6 mice by modulating the cAMP/PKA and cAMP/Epac1 signaling pathways. Biol Pharm Bull. 2022;45(8):1043–52. Epub 2022/05/23. 10.1248/bpb.b21-01057
29 Wang WB, Li JT, Hui Y, Shi J, Wang XY, Yan SG. Combination of pseudoephedrine and emodin ameliorates LPS-induced acute lung injury by regulating macrophage M1/M2 polarization through the VIP/cAMP/PKA pathway. Chin Med. 2022;17(1):19. Epub 2022/02/07. 10.1186/s13020-021-00562-8