
Download
ORIGINAL ARTICLE
Unmasking chronic granulomatous disease: A routine diagnostic workup in a Brazilian children’s hospital
Karina Mescouto de Meloa*, Anna C.S. Diasb, Robéria P. Mendonçab, Cláudia F.C. Valentea, Fabíola S. Tavaresa,c, Agenor C.M. Santos Júniord, Laísa M. Bomfima, Jeane S.R. Martinsa, Ricardo Camargod
aClinic of Allergy and Immunology, Hospital da Criança de Brasília José Alencar, Brasília-Brazil
bFlow Cytometry Service, Translational Research Laboratory, Hospital da Criança de Brasília José Alencar, Brasília-Brazil
cChild and Adolescent Unit, Hospital Universitário de Brasília, Brasília-Brazil
dMolecular Biology, Translational Research Laboratory, Hospital da Criança de Brasília José Alencar, Brasília-Brazil
Abstract
The diagnosis of chronic granulomatous disease (CGD), a congenital immunodeficiency affecting phagocyte function, remains a challenge for patients in Latin America. It is well established that dihydrorhodamine (DHR) flow cytometry is the most commonly used screening assay; however, few pediatric immunology centers in Brazil perform this test. This study reports data from a routine diagnostic workup for CGD conducted at a Brazilian children’s hospital. A three-year prospective study was performed, enrolling children with clinical features suggestive of immunodeficiency who were screened using DHR. Sanger sequencing of the NCF1 (neutrophil cytosolic factor 1) and CYBB (cytochrome b-245, beta chain) genes was conducted in children with two consecutive abnormal DHR results. A total of 255 patients—62% males—with a median age of 3.2 years (range: 1 month–17.8 years) were evaluated. Six patients (2.4%) had abnormal DHR tests, and four of them (1.6%) received a definitive diagnosis of CGD. Most children presented with pneumonia and/or abscesses during the first year of life as the clinical manifestation of CGD. Two of the four diagnosed patients were receiving continuous antibiotics and two underwent transplantation. Pathogenic variants were identified in NCF1 (three cases) and CYBB (one case). The hospital-based diagnostic workup for CGD identified approximately one new case per 60 tested patients, indicating a high frequency of the disease in the study population. This approach may represent a valuable strategy for identifying new pediatric CGD cases in resource-limited settings.
Key words: immunodeficiency, chronic granulomatous disease, rhodamine 123, pediatrics, DNA sequencing
*Corresponding author: Karina Mescouto de Melo, Clinic of Allergy and Immunology, Hospital da Criança de Brasília José Alencar, Brasília-Brazil. Email address: [email protected]
Received 17 February 2025; Accepted 29 June 2025; Available online 1 September 2025
Copyright: de Melo KM, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Chronic granulomatous disease (CGD) is a life-threatening inborn errors of immunity (IEI). Most patients present early in life with severe or recurrent bacterial and fungal infections, primarily affecting the skin, subcutaneous tissues, respiratory tract, lymph nodes, and liver; inflammatory gastrointestinal disorders, susceptibility to granulomas in deep organs, and autoimmune diseases (AIDs) are also frequently observed.1–4
CGD is caused by defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, which is responsible for the respiratory burst in phagocytic leukocytes.1,2 The two most common pathogenic variants associated with CGD in outbred populations are found in the CYBB (cytochrome b-245, beta chain) gene (encoding gp91phox), which is X-linked (accounting for approximately 70% of cases), followed by autosomal recessive mutations in NCF1 (neutrophil cytosolic factor 1) (encoding p47phox), which occur in around 25% of cases.3–7
The dihydrorhodamine (DHR) 123 test by flow cytometry is the preferred method for diagnosing CGD in children, as it is a highly sensitive and a reliable assay that requires only a small blood sample.8–11 The disease-causing variant should be identified in every CGD patient to determine the inheritance pattern, which is critical for genetic counseling, prognosis, and assessing whether a related donor carries the pathogenic variant.5,12,13
CGD has an estimated incidence of approximately 1 in 200,000 live births.3,14,15 However, the true prevalence in Brazil, the most populous country in Latin America, remains unknown.14,16 The primary challenge faced by pediatric immunologists in diagnosing CGD is limited access to specific tests, such as the DHR assay.17 Here, we report a three-year routine diagnostic workup for CGD conducted at a public children’s hospital in Midwest Brazil. Patients were screened using the DHR assay, followed by the Sanger sequencing of CYBB and NCF1. To the best of our knowledge, this is the first study describing a diagnostic strategy for CGD in a Brazilian children’s hospital.
Material and Methods
Patients and samples
Patients aged between 1 month and 18 years were enrolled in the study. All participants exhibited clinical manifestations of CGD according to the European Society for Immunodeficiencies (ESID) criteria,18 which include at least one of the following: deep-seated infections caused by bacteria and/or fungi (e.g., abscesses, osteomyelitis, lymphadenitis); recurrent pneumonia; lymphadenopathy and/or hepatomegaly and/or splenomegaly; obstructing or diffuse granulomas (affecting the gastrointestinal or urogenital tract); or chronic inflammatory manifestations (e.g., colitis, liver abscesses, and fistula formation). Patients with a diagnosis of neoplasia or well-defined syndromes were excluded, and those receiving immunosuppressive drugs or hospitalized in the intensive care unit were included only if they were clinically stable. Informed consent was obtained from all caregivers, and an adapted version of the informed consent was also applied for patients older than 6 years. Patients were evaluated by the same immunology team at a children’s hospital in Brasília, Brazil. The study was approved by the Brazilian Ethical Committee—Plataforma Brasil, under approval number CAAE: 36211220.6.0000.0023. This manuscript reports data collected from September 2021 to August 2024.
Data collection: Clinical data were obtained from medical records using the REDCap® software. Demographic information (age, gender), clinical data (medical history, medications), and laboratory results—including complete blood count, serum immunoglobulin (Ig) levels (IgG, IgM, IgA), and lymphocyte subsets (cluster of differentiation [CD]3, CD4, CD8, CD19, CD16/CD56)—were recorded. Microbiological cultures, histology, and imaging exams were collected for patients with abnormal test results. All experiments were conducted at the Translational Research Laboratory (TLR) of our institution.
Immunophenotyping
An adapted protocol for the DHR assay by flow cytometry, based on previous reports,5,19 was employed in this study. Briefly, two sets of three tubes were prepared for each experiment: blank, stimulated, and unstimulated tubes for both patients and normal controls. One hundred microliters of heparinized venous blood (without prior red cell lysis) diluted in 1x phosphate buffered saline (PBS) was added to all tubes, and 0.8 μL of 1 mM DHR-123 in dimethyl sulfoxide (DMSO) was added to all tubes, except the blank ones. The tubes were then incubated in a shaking water bath at 37°C for 15 minutes. Subsequently, 2 μL of phorbol 12-myristate 13-acetate (PMA) (100 μg/mL) was added to the stimulated tube, followed by an additional 30-minute incubation in the dark. Red cells were lysed using fluorescence-activated cell sorting (FACS) lyse solution and washed twice with 1x PBS buffer. Samples were acquired on a BD FACSCanto® flow cytometer using Diva software, with a minimum of 10,000 neutrophils analyzed per sample. The stimulation index (SI) was calculated by dividing the mean fluorescence intensity (MFI) of the stimulated tube (with PMA) by the MFI of the unstimulated tube (without PMA). Granulocytes were the primary cells of interest (gated using forward scatter [FSC] vs. side scatter [SSC]), while lymphocytes served as a negative control. A healthy donor sample was included in each test as a quality control measure. DHR assay results were interpreted as: SI values ≥ 100 were considered normal, SI values between 60 and 100 were deemed inconclusive, and SI values < 60 were classified as abnormal. The assay was repeated for all cases with inconclusive or abnormal SI values, and a sample was sent for deoxyribonucleic acid (DNA) extraction during the second blood collection.
DNA analysis and sequencing of NCF1 and CYBB
Genomic DNA was extracted from total blood leukocytes using standard procedures. Mutations in the exons and exon-intron boundaries of CYBB and NCF1 were analyzed by polymerase chain reaction (PCR) amplification of each exon, including its intronic boundaries, followed by sequence analysis. Sanger sequencing was performed on PCR products from CYBB (exons 1–13) and NCF1 (exons 2 and 4), using primers described previously.20 Purification of 10 μL of PCR product was conducted using the ExoSAP-IT PCR Product Cleanup Reagent (ThermoFisher Scientific®, Wyman Street, Waltham, MA, USA) at 37°C for 5 minutes. For the sequencing reaction, 3 μL of purified PCR product and 0.3 μM of forward (FWD) or reverse (REV) primers for each target were used, following the manufacturer’s instructions provided with the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies). Sequencing analyses were performed using an ABI3500 Genetic Analyzer® (Life Technologies). Other genes associated with CGD were not analyzed due to cost and technical limitations.
Results
Clinical features of studied patients
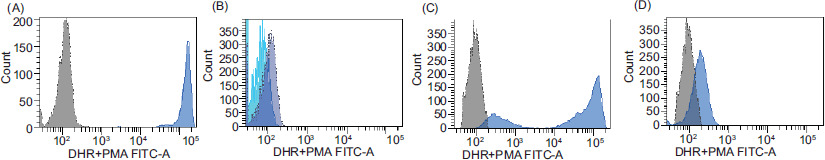
At the end of data collection, a total of 255 patients, including 158 (62.0%) males, were tested for CGD using the DHR assay. The median age of all patients was 3.2 years (range: 1.7 months–17.8 years). As expected, most patients (96.8%) had normal DHR assay results, with a median SI of 367 (range: 1–3373). Two patients had inconclusive results and did not undergo a second round of testing; one patient moved to another country and the other deceased before the repeat test. Six patients (2.4%) had abnormal results upon retesting. Figure 1 illustrates the DHR assay analysis in a healthy control: a patient with X-linked CGD (X-CGD) and his mother who has carrier status (Figure 1).
Figure 1 Neutrophil oxidative burst test using the dihydrorhodamine (DHR) assay. (A) Control sample showing the normal shift of activated neutrophils (blue) compared to unstimulated neutrophils (gray); (B) DHR of a patient with a mutation in CYBB showing the absence of a shift in stimulated neutrophils; (C) DHR of an X-linked carrier of a CYBB defect showing a bimodal population of stimulated neutrophils; (D) DHR of a female patient with autosomal recessive CGD and a defect in NCF1.
Sanger sequencing was performed in six cases. Molecular analysis identified pathogenic variants related to CGD in four patients (1.6%): three in the NCF1 gene (p47phox), including two siblings, and one patient with X-linked CGD (CYBB/gp91phox deficiency).
Targeted sequencing did not reveal any variants in two cases. Patient #53, a 9-year-old girl with a history of recurrent pneumonia, syndromic features, and chronic neurological disease (West syndrome), underwent Whole Exome Sequencing (WES) at a private laboratory. No pathogenic variants related to IEI were identified.
The second patient (P#123, a 16-year-old girl), presented with clinical features of cutaneous and pulmonary mycobacterial disease, accompanied by weight loss and gastrointestinal granulomas. No pathogenic variants were identified in this case either. She was referred for a Next-Generation Sequencing (NGS) panel encompassing 575 genes associated with IEI, including CYBB, CYBA (cytochrome b-245 alpha chain), NCF1, NCF2, NCF4, and RAC2 (Rac family small GTPase 2). Unfortunately, no molecular defect was detected. She received antituberculostatic therapy (isoniazid, rifampin, ethambutol, and pyrazinamide) for a total of 9 months. Despite the lack of a molecular diagnosis, she was started on prophylactic treatment with trimethoprim-sulfamethoxazole (TMP-SMX) and itraconazole based on her clinical diagnosis of CGD. Additionally, she required a course of oral corticosteroids to manage chronic granulomas causing intestinal obstruction. Beyond her clinical challenges, negative social factors, such as addiction to psychoactive substances, significantly impacted her adherence to treatment. Regrettably, the patient passed away at the age of 18 due to sepsis.
Clinical presentation of patients with definitive diagnosis of CGD
The age of clinical presentation among the children with CGD (n = 4) ranged from 1 month to 2 years, with respiratory and cutaneous symptoms being the predominant manifestations. The main clinical and laboratory data of the four patients with a definitive diagnosis of CGD are summarized in Table 1.
Table 1 Clinical and laboratory data of patients with chronic granulomatous disease diagnosed in a Brazilian children’s hospital (2021–2024).
| Patient | P#45 | P#93 | P#204 | P#137 |
|---|---|---|---|---|
| Gender | Female | Male | Male | Male |
| Age (years) | 5.7 | 4.9 | 1.8 | 0.9 |
| Age at onset of symptoms | 2 years | 1 month | 4 months | 2 months |
| First clinical features | PNM | Neonatal sepsis/oral moniliasis | Axillary adenopathy | BCG itis PNM, suppurative lymphadenitis |
| Consanguinity | No | No | No | No |
| Family history | Yes | No | Yes | No |
| Clinical features at diagnosis | PNM, liver and splenic abscesses | Oral moniliasis, PNM, lung abscess | Recurrent diarrhea/adenopathy | BCG itis, skin ulcerous, and adenomegaly |
| Other manifestations | Splenomegaly | Esophageal moniliasis | No | Splenomegaly |
| Medication | TMP-SMX/voriconazole | TMP-SMX | TMP-SMX/itraconazole | TMP-SMX + itraconazole + anti-TB drugs |
| Age (years) | 5.7 | 4.9 | 2.4 | 1 |
| Hemoglobin (g/dL) | 9.6 | 9.1 | 10.6 | 8.1 |
| Neutrophil/μL | 4032 | 4,600 | 4,900 | 9,650 |
| Lymphocytes/μL | 2,500 | 3,700 | 6,100 | 11,020# |
| T CD3+/μL | 2,145 | 3,216 | 3,574.6 | 6,733# |
| T CD4+/μL | 1,617 | 1,029 | 2,000.8 | 1,934 |
| T CD8+/μL | 401 | 2,007 | 1,146.8 | 4,632 |
| CD19+/μL | 175* | 382.8* | 1,781.2 | 3,118 |
| CD16+/CD56+/μL | 87.5 * | 629.2# | 463.6 | 672 |
| IgG (mg/dL) | 1,571# | 1612# | 1,067# | 1,618# |
| IgM (mg/dL) | 324# | 73 | 117 | 158# |
| IgA (mg/dL) | 358# | 493# | 107 | 75 |
| DHR (SI) | 1.81 | 1.03 | 2.54 | 0.72 |
| Isolated microorganism | NI | NI | NI | NI |
| Mutated gene | NCF1 | NCF1 | NCF1 | CYBB |
| Nucleotide change | c.75_76del, exon 2 | c.75_76del, exon 2 | c.75_76del, exon 2 | c.676C>T, exon 7 |
| Current age/follow-up | 9 years/alive transplanted | 7 years 9 months/alive on prophylaxis | 2 years 10 months/alive on prophylaxis | 3 years/alive transplanted |
PNM: Pneumonia, DHR: Dihydrorhodamine, SI: Stimulation index, TMP-SMX: Trimethoprim and Sulfamethoxazole, TB: tuberculosis, NI: Not isolated. *Values lower than Brazilian references, # values above the reference.
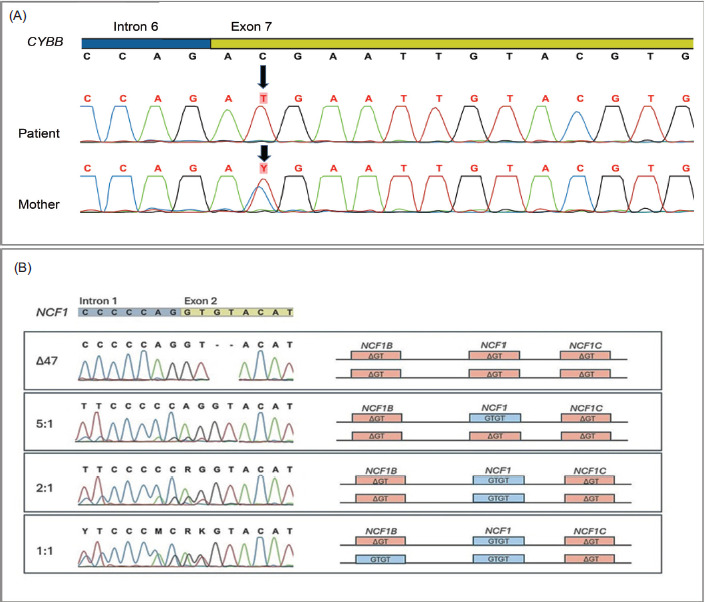
Patient P#137, the only patient with X-linked CGD (X-CGD), experienced four hospital admissions during the first six months of life due to recurrent infections. He underwent multiple courses of antibiotics before being evaluated by a pediatric immunologist. He was referred to our center following an unsuccessful 28-day treatment for cutaneous leishmaniasis. His medical history was consistent with CGD, including recurrent pneumonia, failure to thrive, adverse event to the Bacille Calmette-Guérin (BCG) vaccine, recurrent suppurative lymphadenopathy and splenomegaly. Initial treatment consisted of TMP-SMX combined with itraconazole. However, clinical progression was marked by lymphoproliferative disease, enlargement of axillary lymph nodes (up to 15 × 8 mm), a pulmonary nodule (5 mm), and multiple small splenic abscesses identified on imaging studies. Further evaluation led to a diagnosis of systemic mycobacterial disease secondary to the BCG vaccine. Histological analysis of lymph node tissue revealed granulomas suggestive of mycobacterial infection, although no acid-fast bacilli were observed (Table 1). The patient was treated with a 9-month course of antituberculosis medications, including rifampicin, isoniazid, pyrazinamide, and ethambutol, resulting in clinical improvement. His mother was confirmed to have carrier status (Figures 1 and 2).
Figure 2 Sanger sequencing electropherograms of the CYBB gene (2A) and NCF1 and pseudogenes NCF1B and NCF1C (2B). (A) the first line represents the reference sequence, with the black arrow indicating a single-nucleotide substitution, c.676C>T in exon 7, in an X-CGD patient. The third line shows heterozygosity for the same mutation in the maternal DNA. (B) the first line represents the GTGT sequence configuration in exon 2 of the NCF1 gene and the ΔGT sequences in exon 2 of the pseudogenes NCF1B and NCF1C. The Δ47 sample shows a patient with copies of the NCF1 gene converted from GTGT to ΔGT, indicating the disease condition. The graphical representation elucidates the genomic configurations for each sample, illustrating the presence and ratios of GTGT and ΔGT sequences.
Three patients with defects in NCF1 also experienced severe infections at an early age (Table 1). The only patient with mild symptoms (P#204) was identified due to the diagnosis of his older sister (P#45) during the study.
All patients required hospital admission prior to diagnosis, with a total of 292 cumulative days of hospital stay and an average of 58.4 days per patient. Two patients required intensive care before their diagnosis.
Follow-up
Upon diagnosis, only one patient (P#45) was hospitalized due to an infection. She required a 2-day hospital stay to investigate a pulmonary nodule identified on a chest computed tomography (CT) scan. She was treated with a 60-day course of oral voriconazole for presumed fungal pneumonia, resulting in the resolution of the imaging findings.
Two patients (P#45 and P#137) underwent hematopoietic cell transplantation (HCT) from unrelated donors at the ages of 8 years 2 months and 2 years 10 months, respectively. Both transplanted patients are currently alive, with survival durations of 10 months (P#45) and 3 months (P#137) posttransplantation. Patient P#45 achieved complete engraftment, as confirmed by a normal DHR test following HCT. In contrast, the patient with X-CGD (P#137) demonstrated partial engraftment. He remains on immunosuppressive therapy in combination with isoniazid and SMX-TMP with no significant side effects reported, and continues to be closely monitored by the transplant team at the hospital.
Patients P#93 and P#204 are currently receiving oral prophylaxis with TMP-SMX and itraconazole while awaiting a compatible donor for HCT.
Discussion
The true prevalence of CGD in Brazil remains unknown. Oliveira-Junior et al. (2015) reported 71 cases of CGD in Latin America, with 39% occurring among Brazilian patients.16 Limited access to specific immunologic tests has been identified as a significant barrier to expanding the diagnosis of IEI, such as CGD, in Brazil and across Latin America.17 The primary aim of this study was to describe the results of a 3-year diagnostic workflow for CGD in symptomatic patients treated at a Brazilian hospital. This approach facilitated timely diagnoses and informed clinical decision-making when necessary. To the best of our knowledge, this is the first study to evaluate the routine use of the DHR assay in a pediatric hospital in Brazil.
Currently, the DHR assay is considered the method of choice due to its relative ease of performance, high sensitivity even in detecting very low numbers of functional neutrophils, and its utility in predicting the residual superoxide activity of patient neutrophils, which correlates with survival in individuals with CGD.8,9 Additionally, the assay can distinguish between X-linked CGD, autosomal recessive (AR)-CGD, and X-linked carrier status.6,14
The implementation of the DHR test at an immunology center can be challenging, as it is a functional assay of neutrophils that requires careful consideration of cell viability and the potential for nonspecific neutrophil activation, which may confound the analysis.9 Current recommendations from immunology societies advise considering a minimum of two abnormal results to define a possible case of CGD.18 To minimize false results, all samples in this study were collected on-site and experiments were performed within four hours of blood collection.
It is well established that genetic testing is essential for the definitive classification of CGD, with DHR assay patterns supporting clinical suspicion. In this context, the DHR protocol was complemented by the sequencing of NCF1 and CYBB. In over three years, we confirmed four cases of CGD among 255 tested patients, corresponding to approximately one new case of CGD per 60 patients tested. It is important to note that our cohort was preselected for suspected immunodeficiency, which increases the likelihood of positive findings. Nevertheless, these results underscore the critical role of a screening test for CGD in the study population and its utility in identifying a life-threatening condition in a pediatric hospital setting. Our data also suggest that CGD may be underdiagnosed in our region, as previously highlighted.17 According to the Latin American Society for Immunodeficiencies (LASID) registry website,21 fewer than 500 patients are currently documented as having a diagnosis of CGD. Early diagnosis of CGD offers numerous benefits, including appropriate and tailored management of infections and comorbidities, timely definitive therapeutic interventions, and the identification of affected family members.6,9 Brazil, with its vast geographic area and an estimated population of 200 million, likely harbors many undiagnosed children with CGD if the global prevalence of the disease is taken into account. For countries with limited resources, flow cytometry-based assays such as DHR represent an optimal approach for initial screening and can serve as a stepping stone for targeted sequencing.10 Despite certain limitations of the method, such as challenges in detecting mild mutations or cases where neutrophil function is not entirely absent, this report highlights the value of DHR in uncovering new cases of CGD.
Patients with a definitive diagnosis exhibited the typical clinical features of CGD,2,8,15,16 including one case of BCG-itis, a common manifestation of CGD in Brazil.16 Several patients experienced recurrent infections and nearly all required prolonged hospitalizations to manage these conditions. In the most striking case, the boy with X-linked CGD (P#137) spent more than two months in the hospital during his first year of life prior to diagnosis, resulting in failure to thrive, a well-documented complication of pediatric CGD. Therefore, efforts to establish a timely diagnosis of CGD in our setting not only provide an opportunity for curative interventions but also enable better control of the disease’s clinical manifestations through the implementation of prophylactic measures.
Consanguinity was not observed in our cohort; however, pathogenic variants in NCF1 were identified in 3/4th of the cases. Typically, these variants are associated with a more favorable clinical course due to residual reactive oxygen species (ROS) production by neutrophils, although fatal outcomes can still occur.5,6,22 All pathogenic variants identified in our patients have been previously reported in other studies.4,7,22 Prando-Andrade et al. described two Brazilian siblings with the same variant leading to A47-CGD, as observed in our patients.23 Testing for CGD should be considered in relatives of diagnosed patients, even in the absence of characteristic clinical signs or symptoms. It is plausible that autosomal recessive forms of CGD may occur more frequently than currently documented.
The pathogenic nonsense variant c.676C>T in exon 7 of the CYBB gene identified in patient P#137 has been well documented in previous studies.15,16,24,25 These studies, including those involving Latin American patients, have reported heterogeneous variants within this gene, with no significant predominance of a specific variant and no evidence of a founder effect, as might be expected for a disease associated with a potentially fatal phenotype.16,25
Genetic analysis of CYBA (p22phox), NCF2 (p67phox), NCF4 (p40phox), and CYBC1 (EROS)6,7 was not performed in all cases, representing the main limitation of our study. However, the two patients with abnormal DHR results were further evaluated using additional molecular tests, and no defects were identified.
As reported in other countries,8,10 molecular testing is not widely available in most cities in Brazil and often requires considerable time to yield results, leaving many patients with immunodeficiency features undiagnosed. Although the DHR assay has certain limitations, such as challenges in interpretation and the potential for false positive results, the data from our study demonstrate that implementing the DHR test in a hospital-based setting offers a significant advantage by enabling the rapid identification of potential CGD cases, thereby guiding further targeted testing. Multicenter studies conducted in our country with longer follow-up periods could provide more comprehensive insights into the epidemiology and clinical characteristics of CGD in Brazil.
Conclusion
Here, we report the results of a practical integration of flow cytometry and DNA sequencing assays into the routine diagnostic workup for CGD at a Brazilian children’s hospital. This diagnostic strategy has the potential to reveal the frequency of pediatric CGD in our setting.
Acknowledgments
The authors gratefully acknowledge the staff of the Clinical Analysis Laboratory and physicians from the Infectious Diseases and Allergy and Immunology Units as well as the collaboration of the Teaching and Research Department at our institution.
Author Contributions
All authors contributed equally to this article.
Competing Interests
The authors had no relevant financial interests to disclose.
Conflicts of Interests
The authors declare no conflicts of interest.
Funding
This work was supported by Instituto do Câncer Infantil e Pediatria Especializada (ICIPE), Associação Brasileira de Assistência às Famílias de Crianças Portadoras de Câncer Pedi e Hemopatias (ABRACE).
REFERENCES
1 Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther. 2017;34(12):2543–57. 10.1007/s12325-017-0636-2
2 Staudacher O, Von Bernuth H. Clinical presentation, diagnosis, and treatment of chronic granulomatous disease. Front Pediatr. 2024;12:1384550. 10.3389/fped.2024.1384550
3 Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):89–99. 10.1016/j.hoc.2012.11.002
4 Robles-Marhuenda A, Álvarez-Troncoso J, Rodríguez-Pena R, Busca-Arenzana C, López-Granados E, Arnalich-Fernández F. Chronic granulomatous disease: Single-center Spanish experience. Clin Immunol. 2020;211:108323. 10.1016/j.clim.2019.108323
5 Anjani G, Vignesh P, Joshi V, Shandilya JK, Bhattarai D, Sharma J, et al. Recent advances in chronic granulomatous disease. Genes Dis. 2020;7(1):84–92. 10.1016/j.gendis.2019.07.010
6 O’Donovan CJ, Tan LT, Abidin MAZ, Roderick MR, Grammatikos A, Bernatoniene J. Diagnosis of chronic granulomatous disease: Strengths and challenges in the genomic era. J Clin Med. 2024;13(15):4435. 10.3390/jcm13154435
7 Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–507. 10.1007/s10875-022-01289-3
8 Langer S, Radhakrishnan N, Pradhan S, Das J, Saraf A, Kotwal J. Clinical and laboratory profiles of 17 cases of chronic granulomatous disease in North India. Indian J Hematol Blood Transfus. 2021;37(1):45–51. 10.1007/s12288-020-01316-6
9 Yu JE, Azar AE, Chong HJ, Jongco AM, Prince BT. Considerations in the diagnosis of chronic granulomatous disease. J Pediatr Infect Dis Soc. 2018;7(suppl_1):S6–11. 10.1093/jpids/piy007
10 El Hawary R, Meshaal S, Deswarte C, Galal N, Abdelkawy M, Alkady R, et al. Role of flow cytometry in the diagnosis of chronic granulomatous disease: The Egyptian Experience. J Clin Immunol. 2016;36(6):610–8. 10.1007/s10875-016-0297-y
11 O’Gorman MR, Corrochano V. Rapid whole-blood flow cytometry assay for diagnosis of chronic granulomatous disease. Clin Diagn Lab Immunol. 1995;2(2):227–32. 10.1128/cdli.2.2.227-232.1995
12 Roos D, Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol. 2014;175(2):139–49. 10.1111/cei.12202
13 Roos D. Chronic granulomatous disease. Methods Mol Biol. 2019;1982:531–42. 10.1007/978-1-4939-9424-3_32
14 Lacerda-Pontes R, Gomes LN, Albuquerque RSD, Soeiro-Pereira PV, Condino-Neto A. The extended understanding of chronic granulomatous disease. Curr Opin Pediatr. 2019;31(6):869–73. 10.1097/MOP.0000000000000830
15 Al Kuwaiti AA, Al Dhaheri AD, Al Hassani M, Ruszczak Z, Alrustamani A, Abuhammour W, et al. Chronic granulomatous disease in the United Arab Emirates: Clinical and molecular characteristics in a single center. Front Immunol. 2023;14:1228161. 10.3389/fimmu.2023.1228161
16 De Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PVS, Schimke LF, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: First report from the LASID registry: Chronic granulomatous disease in Latin America. Pediatr Blood Cancer. 2015;62(12):2101–7. 10.1002/pbc.25674
17 Costa-Carvalho B, González-Serrano M, Espinosa-Padilla S, Segundo G. Latin American challenges with the diagnosis and treatment of primary immunodeficiency diseases. Expert Rev Clin Immunol. 2017;13(5):483–9. 10.1080/1744666X.2017.1255143
18 European Society For Immunodeficiencies: Diagnosis criteria. https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria. Accessed November, 2024.
19 Golightly M. Dihydrorhodamine (DHR) flow cytometry test for chronic granulomatous disease (CGD): A simple test for routine clinical flow cytometry. ICCS e-Newsletter. https://www.cytometry.org/newsletters/eICCS-2-1/article6.php. Accessed November 10, 2023.
20 Oliveira Júnior EB, Condino Neto A. Uma nova abordagem para o estudo dos defeitos genético-moleculares da doença granulomatosa crônica e análise de suas relações genótipo-fenótipo [Master’s Thesis]. São Paulo: Instituto de Ciências Biomédicas, USP; 2010.
21 Latin Society For Immunodeficiencies: https://lasidregistry.org/lasid. Accessed: January 1, 2025.
22 Kuhns DB, Hsu AP, Sun D, Lau K, Fink D, Griffith P, et al. NCF1 (p47phox)-deficient chronic granulomatous disease: Comprehensive genetic and flow cytometric analysis. Blood Adv. 2019;3(2):136–47. 10.1182/bloodadvances.2018023184
23 Prando-Andrade C, Agudelo-Florez P, Lopez JA, Paiva MADS, Costa-Carvalho BT, Condino-Neto A. Autosomal chronic granulomatous disease: Case report and mutation analysis of two Brazilian siblings. J Pediatr (Rio J). 2004;80(5):425–8.
24 Abd Elaziz D, El Hawary R, Meshaal S, Alkady R, Lotfy S, Eldash A, et al. Chronic granulomatous disease: A cohort of 173 patients—10 years single center experience from Egypt. J Clin Immunol. 2023;43(8):1799–811. 10.1007/s10875-023-01541-4
25 Rae J, Newburger PE, Dinauer MC, Noack D, Hopkins PJ, Kuruto R, et al. X-linked chronic granulomatous disease: Mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase. Am J Hum Genet. 1998;62(6):1320–31. 10.1086/301874