
Download
RESEARCH ARTICLE
SLC27A3 downregulation restores Th17/Treg balance and alleviates COPD via JAK2/STAT3 pathway inhibition
Xiaoping Li, Ji Liu*, Zehui Jing, Shuxia Li
Department of Geriatric Medicine, Qinghai University Affiliated Hospital, Xining, Qinghai, China
Abstract
The main goal of this investigation is to find out how solute carrier family 27 member 3 (SLC27A3) is expressed in the lung tissue of mice with chronic obstructive pulmonary disease (COPD), and how it relates to lung function. A model of COPD was established by exposing organisms to cigarette smoke, followed by investigating the role of SLC27A3 in COPD through experiments conducted both in living organisms and in laboratory settings. Knockout mice lacking SLC27A3 were produced through siRNA transfection to investigate lung function and inflammatory response, using methods such as hematoxylin-eosin staining and enzyme-linked immunosorbent assay. Western blotting was carried out to analyze the expression of SLC27A3. Naïve CD4+ T-cells were stimulated with anti-CD3, anti-CD28, transforming growth factor (TGF)-β, and/or interleukin (IL)-6, and their differentiation into Th17 or Treg cells was promoted, as assessed by flow cytometry. The pathway expression of JAK2/STAT3 was detected using Western blotting. Mice with COPD that had higher expression levels of SLC27A3 in their lung tissue display abnormalities in lung architecture and function, as well as an imbalance between Th17 and Tregs and an elevated inflammatory response. In COPD mice with SLC27A3 knockdown, the JAK2/STAT3 pathway was repressed, lung inflammation was decreased, Th17/Treg balance was improved, and lung functioning was improved. In conclusion, the findings of this study suggest that downregulating SLC27A3 has the potential to attenuate the inflammatory response, mitigate COPD progression, and rebalance the Th17/Treg ratio by inhibiting the JAK2/STAT3 signaling pathway. These results lay a foundation for utilizing SLC27A3 as a potential therapeutic target to modulate the JAK2/STAT3 pathway for the treatment of COPD, with the aim of enhancing lung function, reducing inflammation, and restoring Th17/Treg equilibrium in a clinical context.
Key words: chronic obstructive pulmonary disease, solute carrier family 27 member 3, JAK2/STAT3, Th17/Treg balance, inflammatory response, cigarette smoke
*Corresponding author: Ji Liu, Department of Geriatric Medicine, Qinghai University Affiliated Hospital, No. 29, Tongren Road, Chengxi District, Xining, Qinghai, China. Email address: [email protected]
Received 10 September 2024; Accepted 26 November 2024; Available online 1 January 2025
Copyright: Li X, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
As one of the most prevalent chronic pulmonary respiratory disorders, chronic obstructive pulmonary disease (COPD) is defined by an irreversible airway obstruction caused by small airway remodeling, emphysema, and chronic bronchitis, as well as an accelerated reduction in lung function.1 Currently, it is widely accepted that aging, environmental influences, particulate matter 2.5 (PM2.5), and cigarette smoke play key roles in the development of COPD.2 Various mechanisms such as inflammation, dysregulation of protease and antiprotease activity, oxidative stress, cellular senescence, and the death of endothelial and epithelial cells have been identified as significant factors in the initiation and advancement of COPD.3 While the precise immunological mechanisms underlying COPD remain elusive, contemporary research has established connections between the pathophysiology and progression of the condition with various immune cells and inflammatory cytokines. These include but are not limited to TGF-β, IL-17, IL-22, as well as neutrophils, macrophages, and CD4+ and CD8+ T lymphocytes.4 Therefore, it is important to investigate the pathophysiology of COPD in detail and identify targets for successful treatment.
The dysregulation of Th17 and regulatory T (Treg) cells is considered a key factor in the pathogenesis of COPD, in spite of the exact mechanisms of the disease remaining unclear.5 In individuals with COPD, there is a shift in the Th17/Treg cell equilibrium toward Th17 cells, leading to heightened inflammation in the lungs and airways, thereby exacerbating alveolar degradation.6 In contrast, Treg cells are a T-cell subtype responsible for maintaining immune homeostasis by suppressing abnormal immune responses and inflammation.7
According to Zhang et al.’s study, which used machine learning algorithms to predict immune infiltration-related diagnostic gene biomarkers in patients with chronic obstructive pulmonary disease, solute carrier family 27 member 3 (SLC27A3) is a diagnostic marker for COPD.8 Because of its crucial role in facilitating the uptake of fatty acids by endothelial cells,9 the membrane protein SLC27A is in focus for its involvement in various diseases, such as autism,10 colon cancer,11 glioblastoma,12 and more. Within the SLC27A3 gene lies the code for FATP3, a protein vital for transporting long-chain fatty acids and triggering the activation of very long-chain fatty acids. Upregulated levels of FATP3 can enhance lipid metabolism, which is intricately associated with the progression of COPD.13 Thus, we deduce that SLC27A might have a major regulatory function in the onset and progression of COPD.
The hypothesis of this study posited that the reduction of inflammation and alleviation of symptoms in chronic obstructive pulmonary disease induced by cigarette smoke could be achieved by restoring the Th17/Treg balance through the knockdown of SLC27A3. This study’s major goals are to: (i) explain how SLC27A3 expression varies in COPD animal models; (ii) verify whether SLC27A3 can treat COPD that is produced by cigarettes in animal models; and (iii) explain how SLC27A3 lung protection works in COPD.
Method
Animal model
The Experimental Animal Care and Ethics Committee at the Qinghai University Affiliated Hospital (Approval No. P-SL-2023201) gave its approval for animal experiments. Six healthy adult C57BL mice, comprising three male and three female mice, aged 8–10 weeks, weighing 18–22 g, were raised under pathogen-free conditions and exposed to 12 hours of light and 12 hours of dark at 18–22°C. The humidity was 50%–60%. The mice were fed a standard diet and had free access to water purchased from Beijing SPF Biotechnology Co., Ltd. For 2 weeks, five days in a row, mice were exposed to three packs of cigarettes (the Furong brand, Hubei China Tobacco Industry Co., Ltd.). They were placed in a ventilated chamber where they were exposed to either sham air or 4% cigarette smoke at a constant flow rate of 1 L/min using a peristaltic pump. Intratracheally, either 5 nmol of SLC27A3siRNA or 5 nmol of nontargeting controlsiRNA in 30 μL of PBS was administered daily throughout the subsequent week. The experiment comprised four groups, each consisting of six mice: control group, COPD group, COPD+siNC group, and COPD+siSLC27A3 group.
Immunohistochemistry
To retrieve antigens for immunohistochemistry, lung tissue sections from mice with COPD and healthy mice were subjected to deparaffinization, rehydration, and subsequently immersed in sodium citrate buffer. After that, slices were serum-blocked and incubated for an additional night at 4°C with primary antibodies against SLC27A3 (ab262939; Abcam) diluted in PBS. Sections were then observed using a microscope imaging system (Nikon, Tokyo, Japan) after being treated with HRP-conjugated secondary antibodies for 1 hour at room temperature.
H&E staining
After being ethanol-dehydrated, lung tissue from mice with COPD and healthy mice was embedded in paraffin. After that, the tissue was divided into 5 μm-thick pieces. For histological assessment, particular sections were stained with hematoxylin (Solarbio, H8070, Beijing, China) and eosin (Sangon, A600190, Shanghai, China) following the dewaxing and rehydration procedure. Next, the ImageJ program was used to observe.
Enzyme-linked immunosorbent assay (ELISA)
Samples of the lungs were taken and stored at −80°C right away. Commercial ELISA kits (Cat. No. 70-EK282/4-96, 70-EK206/3-96, and 70-EK201B/3-96) were utilized to test lung tissue inflammatory factors, specifically TNF-α, IL-6, and IL-β. The kits were used in accordance with the manufacturer’s instructions and were bought from Multisciences (Lianke, Hangzhou, China) Biotechnology Co., Ltd.
Naïve CD4+ T-cell isolation and differentiation
Using the CD4+CD62L+ T-cell isolation kit II (Miltenyi Biotech), naive CD4+ T-cells were extracted from mice lungs. Naked CD4+ T-cells are cultured in vitro with anti-CD3 and anti-CD28 (2 μg/mL) under differentiation conditions for Th17 (TGF-β, 5 ng/mL; IL-6, 20 ng/mL) and Treg (TGF-β, 5 ng/mL). After 3 days of stimulation, the cells were taken out.
Flow cytometry
A cell suspension containing 10^6 cells per milliliter was prepared. Following fixation and permeabilization using 0.1% Triton X, the cells were labeled with PE-conjugated anti-Treg and anti-TH17 antibodies. Subsequently, they were stained with FITC-conjugated anti-CD4 and APC-conjugated anti-CD25 antibodies. Isotype controls were included in all experiments to account for any nonspecific signals. FACS CantoTM II (BD Biosciences, San Jose, CA, USA) was used to evaluate T-cells, and FlowJo7.6.1 software (Tree Star, USA) was used to analyze the results.
Western blot assay
RIPA lysis buffer (Solarbio Life Sciences, Beijing, China) was used to lyse lung tissue on ice. After centrifugation, the proteins were separated and the resulting supernatant was gathered. Total protein was mixed with SDS sample buffer, boiled, separated using SDS-PAGE, and then transferred onto a PVDF membrane, which was subsequently blocked with skimmed milk for 2 hours. Following an overnight incubation at 4°C with specific primary antibodies, the PVDF membrane underwent treatment with an HRP-conjugated secondary antibody for 2 hours after being washed with buffer three times. Finally, an ECL reagent was utilized to visualize the bands on the membrane—SLC27A3 (ab262939, 1:5000, abcom), Foxp3 (1:1000, ab20034, abcom), RORγt (1:1000, ab140807, abcom), JAK2 (1:1000, ab108596, abcom), p-JAK2 (1:1000, ab219728, abcom), STAT3 (1:1000, ab68153, abcom), p-STAT3 (1:1000, ab267373, abcom), and GAPDH (ab9485, 1:1000, abcom).
Statistical analysis
Data analysis was done using GraphPad Prism 8 software (San Diego, CA, USA),. The findings are presented as mean ± SD. A one-way analysis of variance (ANOVA) or a two-sided, unpaired Student’s t-test was used for statistical comparisons, and when necessary, Dunnett’s test or Tukey’s post hoc test was used. A p-value of less than 0.05 was considered to be statistically significant.
Result
SLC27A3 expression is elevated in COPD models
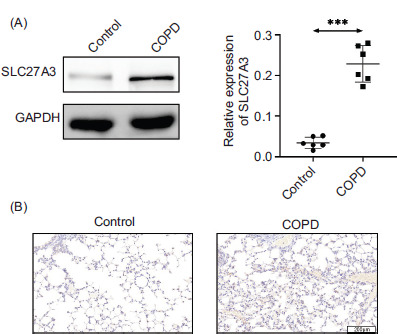
To investigate the potential role of SLC27A3 in the development of COPD, we examined the expression of SLC27A3 in the lung tissues of COPD mice from both control and experimental groups. The results from Western blot analysis revealed that the experimental group of COPD mice exhibited elevated levels of SLC27A3 in their lung tissues compared to the mice exposed to ambient air (Figure 1A). Furthermore, lung tissues from mic with COPD have elevated levels of SLC27A3, as demonstrated by immunohistochemistry staining (Figure 1B). Therefore, it appears that SLC27A3 is involved in the initiation and progression of COPD.
Figure 1 SLC27A3 expression is elevated in COPD models. (A) Protein expression of SLC27A3 in lung tissue. (B) Immunohistochemical staining of SLC27A3 in lung tissue. Values are presented as mean ± SD. ***p < 0.001 versus control group. n=6.
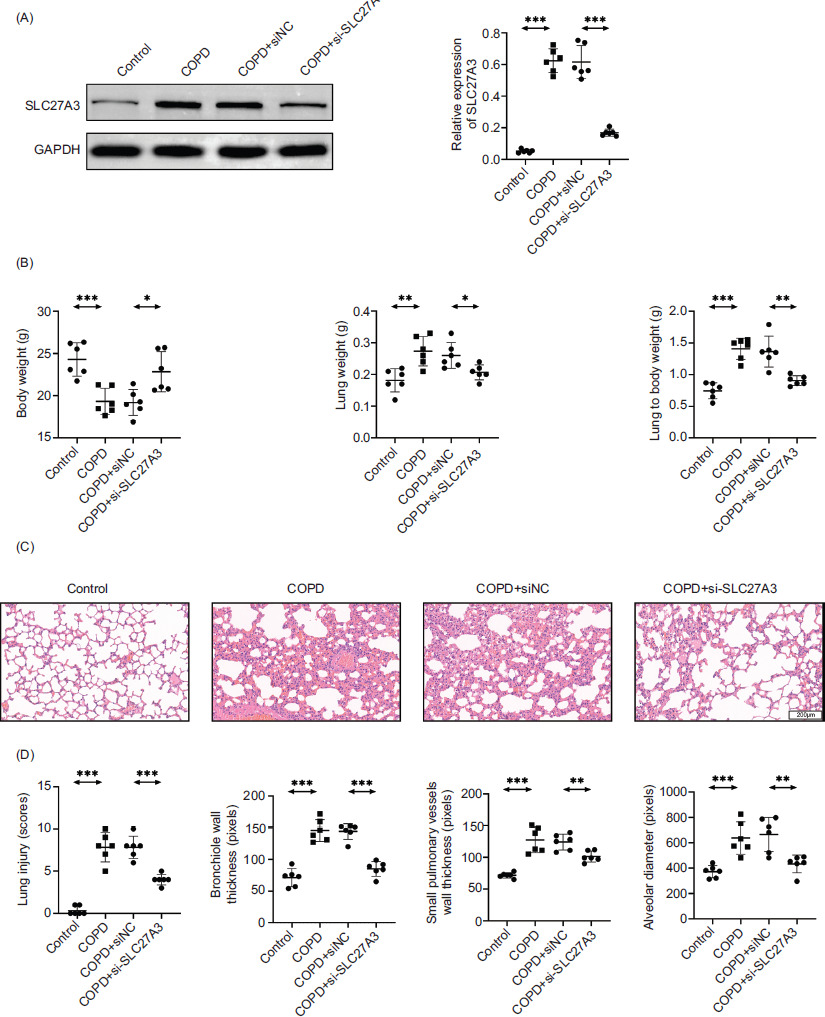
Knockdown of SLC27A3 improves lung injury in COPD mice
To confirm that SLC27A3 is relevant to COPD, we knocked it down in mice. The results of Western blotting verified that we were able to effectively knock down SLC27A3 (Figure 2A). The weights of mouse bodies, lung tissues, and lung indices were quantified, and the findings substantiated that the suppression of SLC27A3 leads to the amelioration of COPD symptoms (Figure 2B). By conducting H&E staining analysis on lung tissues (Figure 2C), it was revealed that the suppression of SLC27A3 led to enhancements in lung damage scores, reductions in small pulmonary vessel wall thickness, alleviation of bronchiolar stenosis, and decreases in alveolar diameter. These factors were previously compromised by exposure to cigarette smoke (Figure 2D). The findings demonstrated that SLC27A3 is necessary for the onset of COPD and that preventing it greatly lessened the severity of COPD.
Figure 2 Knockdown of SLC27A3 improves lung injury in COPD mice. (A) Protein expression of SLC27A3 in lung tissue. (B) Mouse weight, lung weight, and lung index. (C) H&E staining to detect pathological changes of lung tissue in each group. (D) Statistics of lung injury scores, bronchial wall thickness, small pulmonary vessels wall thickness, and alveolar diameter. Values are presented as mean ± SD. *p < 0.01, **p < 0.01, ***p < 0.001. n=6.
Knockdown of SLC27A3 inhibits inflammation in COPD mice
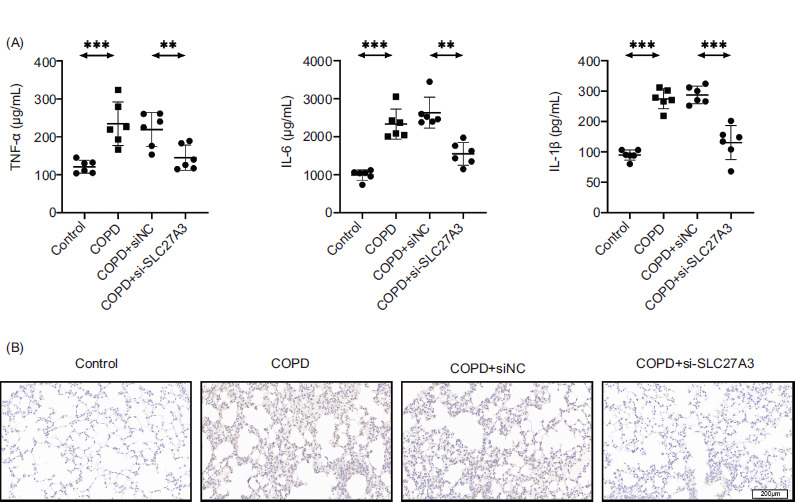
Next, we looked at how SLC27A3 affected the inflammation in COPD mice. Based on the findings from an ELISA study, it was observed that suppressing SLC27A3 helped reduce the harmful effects of cigarette smoke on the levels of IL-6, IL-1β, and TNF-α (Figure 3). These results indicate the essential role of SLC27A3 in inflammatory responses associated with COPD.
Figure 3 Knockdown of SLC27A3 inhibits inflammation in COPD mice. (A) ELISA to detect the levels of inflammatory factors (IL-6, IL-1β, TNF-α) in serum. (B) Immunohistochemical determination of TNF-α expression in lung tissue. Values are presented as mean ± SD. **p < 0.01, ***p < 0.001. n=6.
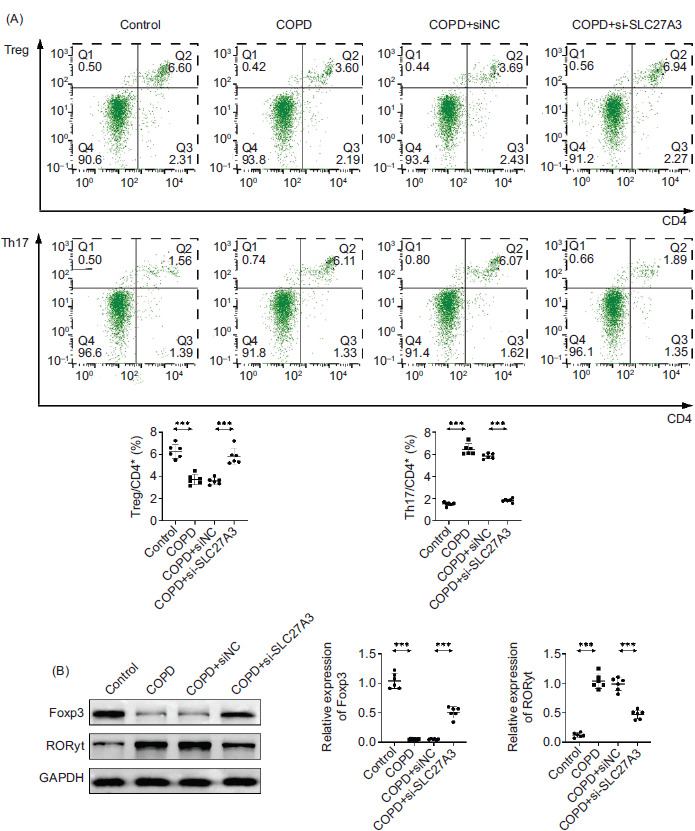
Knockdown of SLC27A3 restores Th17/Treg balance in COPD mice
One notable characteristic of inflammatory disorders is the disrupted Th1/Th17 reaction, frequently coupled with a reduction in Treg cell levels. While Th17 cells contribute to inflammation when in excess, Treg cells act as suppressors, with their imbalance potentially exacerbating the development of inflammatory conditions. Therefore, maintaining the equilibrium between Th17 and Treg cells is important for managing COPD.14 We further investigated the impact of SLC27A3 knockdown on Th17/Treg balance in the lungs of COPD mice to confirm the effect of SLC27A3 on COPD animals. The number of TH17 cells was increased and the percentage of Treg cells was considerably decreased by cigarette smoke (Figure 4A). Downregulating SLC27A3 can reverse the increase in Th17 cells and decrease in Treg cells induced by BYF. In addition, this approach can mitigate the promotion of RORγt expression by cigarette smoke and its suppressive impact on Foxp3 (Figure 4A, B). These findings imply that SLC27A3 knockdown can control the ratio of Th17/Treg cells in COPD mice.
Figure 4 Knockdown of SLC27A3 restores Th17/Treg balance in COPD mice. (A) Flow cytometry was used to detect the proportion of Treg and Th17 cells. (B) Western blotting to detect Foxp3 and RORγt protein expression. Values are presented as mean ± SD. ***p < 0.001. n=6.
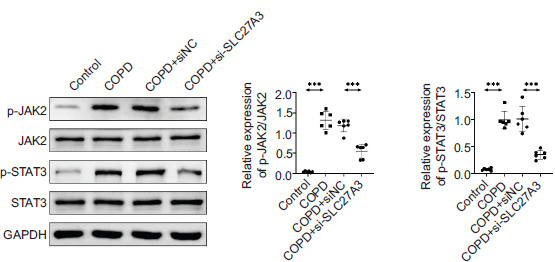
Knockdown of SLC27A3 inhibits the JAK2/STAT3 pathway
Western blotting was utilized to analyze the protein expression levels of JAK2, p-JAK2, STAT3, and p-STAT3 in the lung tissue. Research findings suggest that the phosphorylation levels of JAK2 and STAT3 are suppressed in the context of COPD when SLC27A3 is knocked down (Figure 5). SLC27A3 is thought to stimulate the JAK2/STAT3 pathway in COPD.
Figure 5 Knockdown of SLC27A3 inhibits the JAK2/STAT3 pathway. Western blotting to detect the expression of JAK2, p-JAK2, STAT3, and p-STAT3 proteins. Values are presented as mean ± SD. ***p < 0.001. n=6.
Discussion
The most prevalent chronic respiratory disease, COPD, poses a major hazard to human health due to its high prevalence, disability, and mortality.15 Currently, there is no established curative remedy for COPD. As COPD progresses, patients frequently suffer from systemic complications, leading to malnutrition and impaired muscle function in their extremities and respiratory system. The prevalence of dyspnea is on the rise, leading to a decrease in exercise capacity and quality of life, which poses a significant burden on society.16,17 As a result, it is crucial to identify efficient treatment methods for COPD and clarify its mechanisms of action.
Around 80% of individuals with COPD have a background of smoking, which is identified as a primary risk element for the development of the condition.18 Smoke containing harmful particles causes inflammation of the airways, which is made worse for people with COPD. In addition to being a social issue, smoking is harmful to the human body.19 Multiple research studies have shown the effectiveness of quitting smoking in preventing COPD, postponing airflow limitation, and decelerating the decline of lung function.20 A mouse model of COPD induced by cigarette smoke was established in this research. Lung function tests were employed to validate, consistent with prior research, the damaging impact of cigarette smoke on lung function. In COPD mice, cigarette smoke alters the Th17/Treg balance, enhances lung tissue inflammation, and modifies lung tissue morphology.
The association between COPD and systemic inflammation, alongside persistent inflammation in the lung tissue and air passages, is well-established. This inflammation is further aggravated during acute exacerbations. Addressing chronic inflammation is a primary therapeutic goal for individuals with COPD.21 Characterized by an increase in (chemotactic) cytokines like TNF-α, IL-6, IL-1β, and similar substances, as well as the presence of inflammatory cells such as neutrophils, macrophages, and lymphocytes in the lungs, this chronic inflammation is easily identifiable.22,23 The results of this study suggest that SLC27A3 may play a role in COPD as it can limit the levels of inflammatory factors such as TNF-α, IL-1β, and IL-6.
The imbalance between Th17 and Treg is crucial to the onset and course of COPD.24 An imbalanced Th1/Th17 immune response plays a pivotal role in inflammatory conditions, often coinciding with a reduction or alteration in Treg cells. The inflammatory role of Th17 cells can worsen inflammatory diseases when they multiply. In contrast, Treg cells exhibit suppressive qualities and can trigger the same diseases when their regulation is disturbed.25 Another reason why SLC27A3 affects COPD could be attributed to the discovery in this study that reducing the protein led to a decrease in Th17 cell differentiation and an increase in Treg cell differentiation in vitro.
It is widely recognized that oxidative stress, apoptosis, and inflammation have been shown to contribute to the progression of COPD through the JAK2/STAT3 pathway, an essential intracellular signaling mechanism.26 In addition, Wang et al. highlighted that the activation of the JAK2/STAT3 pathway could lead to lung epithelial cell death and provoke an inflammatory reaction.27 This work created a mouse model of COPD, and the findings further confirmed that SLC27A3 knockdown can prevent the JAK2/STAT3 pathway from being activated, enhance pulmonary function in COPD mice, and contribute to the onset and progression of COPD.
There are limitations to this study. COPD, a complex disease, is affected by both genetic and environmental factors in its development. It is a diverse syndrome where the extent of lung parenchymal destruction (emphysema) and airway disease can vary among patients with COPD. Genetic determinants may influence heterogeneity and susceptibility to COPD and thus influence experimental outcomes.28
Conclusion
In summary, the results of our research indicate that there is an increase in SLC27A3 expression in animal models with COPD, and that reducing SLC27A3 levels can improve lung condition, decrease inflammation, and rebalance the Th17/Treg ratio in COPD mice. This relationship may be attributed to the inhibition and interference with the JAK2/STAT3 signaling pathway. These findings offer a promising avenue for treating COPD patients through the development of a new, more effective treatment strategy and the clarification of its potential mechanism of action. This may serve as a basis for using SLC27A3 to modulate the JAK2/STAT3 pathway to target COPD and improve lung injury, inflammation, and Th17/Treg balance in clinical settings.
Acknowledgements
Not applicable.
Authors Contribution
All authors contributed to the study conception and design. Material preparation and the experiments were performed by Xiaoping Li. Data collection and analysis were performed by Ji Liu and Zehui Jing. The first draft of the manuscript was written by Shuxia Li, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article. The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors state that there are no conflicts of interest to disclose.
Ethics Approval
Ethical approval was obtained from the Ethics Committee of Qinghai University Affiliated Hospital (Approval No. P-SL-2023201). The animal experiment complies with the ARRIVE guidelines and is in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
Funding
This work was supported by the Qinghai Provincial Health Committee Key Topic (Grant No. 2023-wjzd-10) and rovincial Key Clinical Specialty Construction Project of Qinghai Province (Grant No.2024 [23]).
REFERENCES
1 Yu H, Lin Y, Zhong Y, Guo X, Lin Y, Yang S, et al. Impaired AT2 to AT1 cell transition in PM2.5-induced mouse model of chronic obstructive pulmonary disease. Respir Res. 2022;23(1):70. 10.1186/s12931-022-01996-w
2 Lu Z, Van Eeckhoutte HP, Liu G, Nair PM, Jones B, Gillis CM, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;204(6):667–81. 10.1164/rccm.202009-3442OC
3 Song H, Jiang L, Yang W, Dai Y, Wang Y, Li Z, et al. Cryptotanshinone alleviates lipopolysaccharide and cigarette smoke-induced chronic obstructive pulmonary disease in mice via the Keap1/Nrf2 axis. Biomed Pharmacother. 2023;165:115105. 10.1016/j.biopha.2023.115105
4 Villasenor-Altamirano AB, Jain D, Jeong Y, Menon JA, Kamiya M, Haider H, et al. Activation of CD8(+) T cells in chronic obstructive pulmonary disease lung. Am J Respir Crit Care Med. 2023;208(11):1177–95. 10.1164/rccm.202305-0924OC
5 Lourenco JD, Ito JT, Martins MA, Tiberio I, Lopes F. Th17/Treg imbalance in chronic obstructive pulmonary disease: Clinical and experimental evidence. Front Immunol. 2021;12:804919. 10.3389/fimmu.2021.804919
6 Thomas R, Qiao S, Yang X. Th17/Treg imbalance: Implications in lung inflammatory diseases. Int J Mol Sci. 2023;24(5). 10.3390/ijms24054865
7 Silva LEF, Lourenco JD, Silva KR, Santana FPR, Kohler JB, Moreira AR, et al. Th17/Treg imbalance in COPD development: Suppressors of cytokine signaling and signal transducers and activators of transcription proteins. Sci Rep. 2020;10(1):15287. 10.1038/s41598-020-72305-y
8 Zhang Y, Xia R, Lv M, Li Z, Jin L, Chen X, et al. Machine-learning algorithm-based prediction of diagnostic gene biomarkers related to immune infiltration in patients with chronic obstructive pulmonary disease. Front Immunol. 2022;13:740513. 10.3389/fimmu.2022.740513
9 Viraragavan A, Willmer T, Patel O, Basson A, Johnson R, Pheiffer C. Cafeteria diet induces global and Slc27a3-specific hypomethylation in male Wistar rats. Adipocyte. 2021;10(1):108–18. 10.1080/21623945.2021.1886697
10 Maekawa M, Iwayama Y, Ohnishi T, Toyoshima M, Shimamoto C, Hisano Y, et al. Investigation of the fatty acid transporter-encoding genes SLC27A3 and SLC27A4 in autism. Sci Rep. 2015;5:16239. 10.1038/srep16239
11 Niculae AM, Dobre M, Herlea V, Vasilescu F, Ceafalan LC, Trandafir B, et al. Lipid handling protein gene expression in colorectal cancer: CD36 and targeting miRNAs. Life (Basel). 2022;12(12). 10.3390/life12122127
12 Korbecki J, Kojder K, Jezewski D, Siminska D, Tomasiak P, Tarnowski M, et al. Reduced expression of very-long-chain Acyl-CoA synthetases SLC27A4 and SLC27A6 in the glioblastoma tumor compared to the peritumoral area. Brain Sci. 2023;13(5). 10.3390/brainsci13050771
13 Pecka-Kielb E, Kowalewska-Luczak I, Czerniawska-Piatkowska E, Zielak-Steciwko AE. Effects of single nucleotide polymorphisms in the SLC27A3 gene on the nutritional value of sheep milk. Animals (Basel). 2020;10(4). 10.3390/ani10040562
14 Xu YQ, Lv W, Wu HJ, Shi S. Ginsenoside regulates Treg/Th17 cell ratio and inhibits inflammation to treat COPD. Pharmazie. 2020;75(11):590–4.
15 Uwagboe I, Adcock IM, Lo Bello F, Caramori G, Mumby S. New drugs under development for COPD. Minerva Med. 2022; 113(3):471–96. 10.23736/S0026-4806.22.08024-7
16 Kahnert K, Jorres RA, Behr J, Welte T. The diagnosis and treatment of COPD and its comorbidities. Dtsch Arztebl Int. 2023;120(25):434–44. 10.3238/arztebl.m2023.0027
17 Guo P, Li R, Piao TH, Wang CL, Wu XL, Cai HY. Pathological mechanism and targeted drugs of COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1565–75. 10.2147/COPD.S366126
18 Fu YS, Kang N, Yu Y, Mi Y, Guo J, Wu J, et al. Polyphenols, flavonoids and inflammasomes: The role of cigarette smoke in COPD. Eur Respir Rev. 2022;31(164). 10.1183/16000617.0028-2022
19 Kaur M, Chandel J, Malik J, Naura AS. Particulate matter in COPD pathogenesis: An overview. Inflamm Res. 2022;71(7-8):797–815. 10.1007/s00011-022-01594-y
20 Su X, Wu W, Zhu Z, Lin X, Zeng Y. The effects of epithelial-mesenchymal transitions in COPD induced by cigarette smoke: An update. Respir Res. 2022;23(1):225. 10.1186/s12931-022-02153-z
21 Cheng Q, Fang L, Feng D, Tang S, Yue S, Huang Y, et al. Memantine ameliorates pulmonary inflammation in a mice model of COPD induced by cigarette smoke combined with LPS. Biomed Pharmacother. 2019;109:2005–13. 10.1016/j.biopha.2018.11.002
22 Wang Y, Liao S, Pan Z, Jiang S, Fan J, Yu S, et al. Hydrogen sulfide alleviates particulate matter-induced emphysema and airway inflammation by suppressing ferroptosis. Free Radic Biol Med. 2022;186:1–16. 10.1016/j.freeradbiomed.2022.04.014
23 Pan D, Ye H, Wang J, Mao J. Gentianine facilitates proliferation and inhibits inflammation and oxidative stress in caerulein-triggered acute pancreatitis cell model. Signa Vitae 2024;20(5).
24 Zhao P, Liu X, Dong H, Tian Y, Feng S, Zhao D, et al. Bufei Yishen formula restores Th17/Treg balance and attenuates chronic obstructive pulmonary disease via activation of the adenosine 2a receptor. Front Pharmacol. 2020;11:1212. 10.3389/fphar.2020.01212
25 Lopes F, Tiberio I, Leme A, Fairclough L. Editorial: The importance of Th17/Treg imbalance in asthma and COPD development and progression. Front Immunol. 2022;13:1025215. 10.3389/fimmu.2022.1025215
26 Milara J, Ballester B, de Diego A, Calbet M, Ramis I, Miralpeix M, et al. The pan-JAK inhibitor LAS194046 reduces neutrophil activation from severe asthma and COPD patients in vitro. Sci Rep. 2022;12(1):5132. 10.1038/s41598-022-09241-6
27 Wang L, Yuan N, Li Y, Ma Q, Zhou Y, Qiao Z, et al. Stellate ganglion block relieves acute lung injury induced by severe acute pancreatitis via the miR-155-5p/SOCS5/JAK2/STAT3 axis. Eur J Med Res. 2022;27(1):231. 10.1186/s40001-022-00860-3
28 Silverman EK. Genetics of COPD. Annual review of physiology. 2020;82(1):413–31.