
Download
ORIGINAL ARTICLE
Shengmai Powder regulates alveolar macrophage PPAR-γ and improves the chronic inflammatory state of chronic obstructive pulmonary disease
Dongmei Liu1, Zongwei Liu1, Xunxun Ma1, Shengjie Wang2, Jie Lin1, Xiuyan Shi1, Xiaoyong Xu3*
1Department of Respiratory Medicine, Lianyungang TCM Hospital Affiliated to Nanjing University of Chinese Medicine, Lianyungang, Jiangsu, China
2Department of Basic Medicine, Kangda College of Nanjing Medical University, Lianyungang, China
3Department of Pulmonary and Critical Care Medicine, The Fourth Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China
Abstract
This study examines the therapeutic effects of Shengmai Powder (SMP) on both in vitro and in vivo models of chronic obstructive pulmonary disease (COPD) and the underlying mechanisms. Cigarette smoke and cigarette extracts were used to create in vitro and in vivo models of COPD. ELISA was used to measure the levels of pro-inflammatory factors (IL-6, TNF-α, and IL-1β) in mouse lung tissue and alveolar macrophages. Flow cytometry assessed the phagocytic capacity of alveolar macrophage. Western blotting was used to analyze the expression of RhoA, PPARγ, IκBα, p-IκBα, P65, and p-P65 in alveolar. The results show that SMP reversed the increased levels of pro-inflammatory factors (IL-6, TNF-α, and IL-1β) in mouse lung tissue and alveolar macrophages induced by cigarette smoke and cigarette extract. SMP also restored the decreased fluorescence intensity and RhoA levels in alveolar macrophages caused by cigarette extract. Additionally, SMP increased PPARγ expression and decreased IκBα and P65 phosphorylation in alveolar macrophages exposed to cigarette extract. Also, the effects of SMP were reversed by PPARγ inhibitors. The study concluded that SMP regulates alveolar macrophage phagocytic function through the PPAR-γ/NF-κB pathway, thereby improving the chronic inflammatory state of COPD.
Key words: alveolar macrophages, COPD, inflammation, NF-κB, phagocytosis, PPARγ, SMP
*Corresponding author: Xiaoyong Xu, Department of Pulmonary and Critical Care Medicine, The Fourth Affiliated Hospital of Nanjing Medical University, 298 Nanpu Road, Nanjing, China. Email address: [email protected]
Received 24 May 2024; Accepted 2 July 2024; Available online 1 September 2024
Copyright: Liu D, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
With a high morbidity and mortality rate, chronic obstructive pulmonary disease (COPD) is both prevalent and frequently occurring. Given the financial costs associated with COPD, it has emerged as a significant public health issue.1 Studies demonstrate that cigarette smoke is a major risk factor for COPD.2 Abnormal inflammatory response, airway blockage, alveolar deterioration, and apoptosis are hallmarks of COPD. In COPD patients, there is increased pulmonary vascular endothelial cell apoptosis and an amplified inflammatory response. These factors exacerbate changes in lung tissue morphology changes, lead to significant anomalies in lung function indicators, and thus impair overall lung function.3,4 The anti-inflammatory properties of local glucocorticoids are the mainstay of current therapy options for chronic pulmonary inflammation. However, due to glucocorticoids’ suppression of local inflammation, local immunity will also be reduced, in which can lead to new infections and inflammation. Additionally, the effects of glucocorticoids on local inflammation can have an inhibitory effect on macrophage healing. These factors may contribute to the low clinical efficacy of glucocorticoid therapy.5
Alveolar macrophages (AM), the most prevalent immune cells in the lungs, are essential for lung homeostasis, tissue repair, and host defense. Dysfunction of AM may contribute to the etiology of COPD.6 When exposed to cigarette smoke extract, the phenotype and function of AM are altered, their exocytotic activity is inhibited, and their phagocytosis rate is significantly reduced.7 Therefore, improving AM’s phagocytic function is also a promising method for treating COPD.
In recent years, traditional Chinese medicine has gained growing importance in illness treatment. The integration of Western medicine and traditional Chinese medicine has produced notable results in managing COPD. This combined approach offers clear benefits over using Western medicine alone, providing a more comprehensive treatment strategy.8 The ingredients of Shengmai Powder (SMP), as given in “The Origin of Medicine”, are: 9 g of Ophiopogon japonicus, 9 g of ginseng, and 6 g of Schisandra -chinensis.9 By inhibiting MAPK signaling, the improved new SMP ameliorates myocardial fibrosis in rats with heart failure.10 Additionally, the relative expression of IL-1β and IL-1R in patients’ serum is positively correlated with SMP, suggesting that SMP is useful in reducing the clinical symptoms of pulmonary tuberculosis.11 However, the role of SMP in COPD remains unclear.
This study aimed to explore whether SMP could decrease chronic inflammation in COPD by regulating alveolar macrophage phagocytic function through the PPAR-γ pathway.
Method
Animal feeding
C57BL/6 mice in good health (4 sets of 6 mice each, 50% male and 50% female), aged 8–10 weeks, weighing 18–22 g, with a light-dark cycle of 12 h, a temperature of 18°C –22°C, and a humidity of 50%–60%. Mice were obtained from Beijing Huafukang Biotechnology Company. This study was approved by the Animal Ethics Committee of Lianyungang TCM Hospital with the Standards for the Care and Use of Laboratory Animals. Mice were caged in a rectangular box containing cigarettes (Furong brand, Hubei China Tobacco Industry Co., Ltd.) in a homemade tobacco smoke inhalation exposure device (30x40x90 cm plexiglass cuboid). The control group was fed normally and in a smoke-free atmosphere. The apparatus exposed the model group mice to cigarette smoke four times a day. Each exposure involved lighting six cigarettes and allowing them to burn for 1 h. The exposure regimen was conducted 6 days a week for 4 weeks.12
Ophiopogon japonicus (9 g), ginseng (9 g), and Schisandra chinensis (6 g) were refluxed and extracted three times (1:10). After filtration, aqueous extracts were concentrated into a fixed amount for intragastric administration.
The experiment was divided into four groups (n=6): control group; COPD group; COPD+8.2 g/kg SMP group; COPD+8.2 g/kg SMP +1 mg/kg GW9662 (Sigma-Aldrich, USA) group.
Cell culture and treatment
Mouse alveolar macrophages (MH-S) from the Bernard Culture Collection in China were cultured in RPMI1640 medium in an incubator at 37°C and 5% CO2. The medium was supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. For exposure experiments, cigarette were lit and smoke was introduced using a vacuum pump. The smoke was inhaled into 50 mL of PBS for 3–5 min. The pH of the resulting cigarette smoke extract (CSE) was adjusted to 7.4 and sterilized using a 0.22 μm micropore needle filter, producing a 100% CSE solution. Cells were then stimulated with 5% CSE for 24 h, followed by 24-hour administration period.13
The experiment was divided into four groups (n=3): control group; CSE group; CSE +200 µg/mL SMP group; COPD + 200 µg/mL SMP + 200 µg/mL GW9662 group.
Enzyme-linked immunosorbent assay (ELISA)
Lung tissue and cell samples were obtained according to manufacturer’s instructions. The levels of inflammatory factors (TGF-α, IL-6, and IL-1β) were measured using an ELISA kit (Lianke Biotechnology Co., Ltd., Hangzhou, China).
Flow cytometry to detect phagocytosis
For 6 h without light, 1 × 105 cells/well were subjected to 0.04 mg/mL fluorescein isothiocyanate (FITC)-labeled E. coli (Invitrogen). Following this, 4% trypan blue (Invitrogen) was added to each well to quench the extracellular fluorescence of FITC-labeled E. coli. Flow cytometry (Becton Dickinson Co., USA) was then used to measure the mean fluorescence intensity (MFI). Higher MFI and increased phagocytosis percentage indicate greater phagocytic capacity.
F-actin ring staining
Cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then treated with 0.25% Triton X-100 in phosphate-buffered saline (PBS). Actin was stained using rhodamine-conjugated phalloidin (1:1000; Cytoskeleton, Inc. Fluorescence images were acquired using a Leica DM2500 microscope (Leica Microsystems GmbH).
Western blot
Cells were lysed using RIPA buffer (Solarbio, Beijing, China), and protein concentration was measured using the BCA technique (Beyotime, China). SDS-PAGE was used to resolve each protein sample before transferring the proteins to a PVDF membrane. The membrane was blocked using 5% BSA for 1 h. Following blocking, the PVDF membrane was incubated with primary antibodies overnight at 4°C with gentle shaking. After washing with TBST, the membrane was incubated with horseradishase-conjugated goat anti-rabbit IgG secondary antibody (1:3000, ZSGB-BIO, China) at room temperature for 60 min. After washing in TBST, color development is done using ECL reagent, and the membrane was scanned and processed. Relative expression levels of each protein were calculated using GADPH as an internal reference, and band grayscale was examined to assess protein expression.
The primary antibodies used were as follows: RhoA (1:1000, ab187027, Abcam, UK), peroxisome proliferator--activated receptor-γ (PPARγ, 1:1000, ab178860, Abcam, UK), IkappaBalpha (IκBα, 1:1000, ab230341, Abcam, UK), p-IκBα (1:1000, ab183503, Abcam, UK), p65 (1:1000, ab32536, Abcam, UK), p-p65 (1:1000, ab6503, Abcam, UK), and GAPDH (1:1000, ab8245, Abcam, UK).
Statistical analysis
Data analysis was performed using GraphPad Prism 9.0 and SPSS 21.0. Data are presented as mean ± standard deviation. Each experiment was conducted with a minimum of three independent replicates. One-way or two-way ANOVA was used to compare means across multiple groups, while the unpaired Student's t-test was used to compare two independent groups. A P-value of <0.05 was considered statistically significant.
Result
SMP reduces inflammation in COPD mice
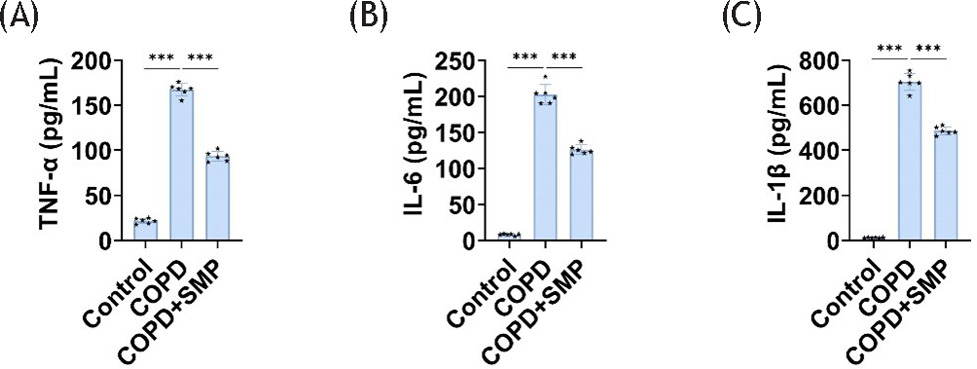
Pulmonary inflammation is the most basic pathological characteristic of COPD. We first examined inflammation in mouse lung tissue. Compared to the control group, levels of inflammatory factors (IL-6, TNF-α, and IL-1β) were significantly elevated in the lung tissue of mice exposed to cigarette smoke. However, the inflammatory factor levels in the lung tissue of mice exposed to SMP were significantly reduced, reversing the negative effects of cigarette smoke (Figure 1A–C). This proves that SMP can reduce chronic airway inflammation in COPD mice.
Figure 1 SMP reduces inflammation in COPD mice. ELISA detects TNF-α (A), IL-6 (B), and IL-1β (C) levels in lung tissue. Data are presented as mean ± SD. *** P < 0.001 versus the control group or COPD group. n=6.
SMP enhances AM’s anti-inflammatory function
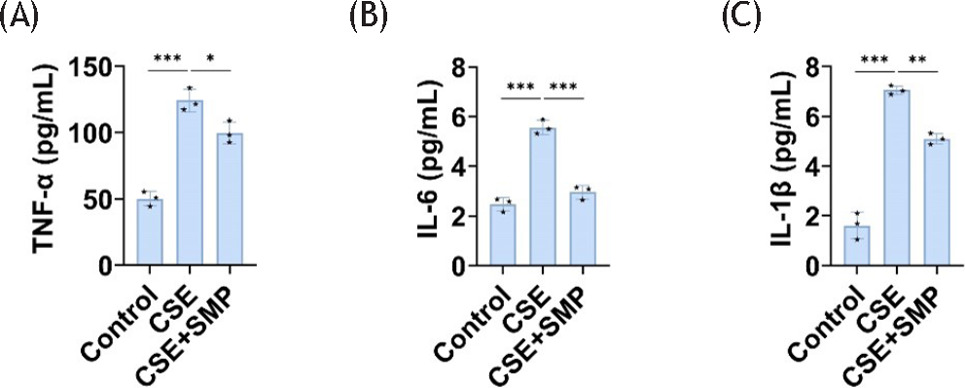
In COPD mice, AM exhibits increased production of inflammatory cytokines.14 MH-S cells were then exposed to 5% CSE for 24 h and inflammatory factors were measured. Compared to the control group, the levels of cellular inflammatory factors (IL-6, TNF-α, and IL-1β) in the CSE group increased significantly, and treatment with SMP reversed this effect (Figure 2A–C). This indicates that SMP enhances anti-inflammatory effects of AM in in vitro models of COPD.
Figure 2 SMP enhances AM’s anti-inflammatory properties. ELISA detects TNF-α (A), IL-6 (B), and IL-1β (C) levels in cells. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, *** P < 0.001 versus the control group or CSE group. n = 3.
SMP enhances AM’s phagocytosis function
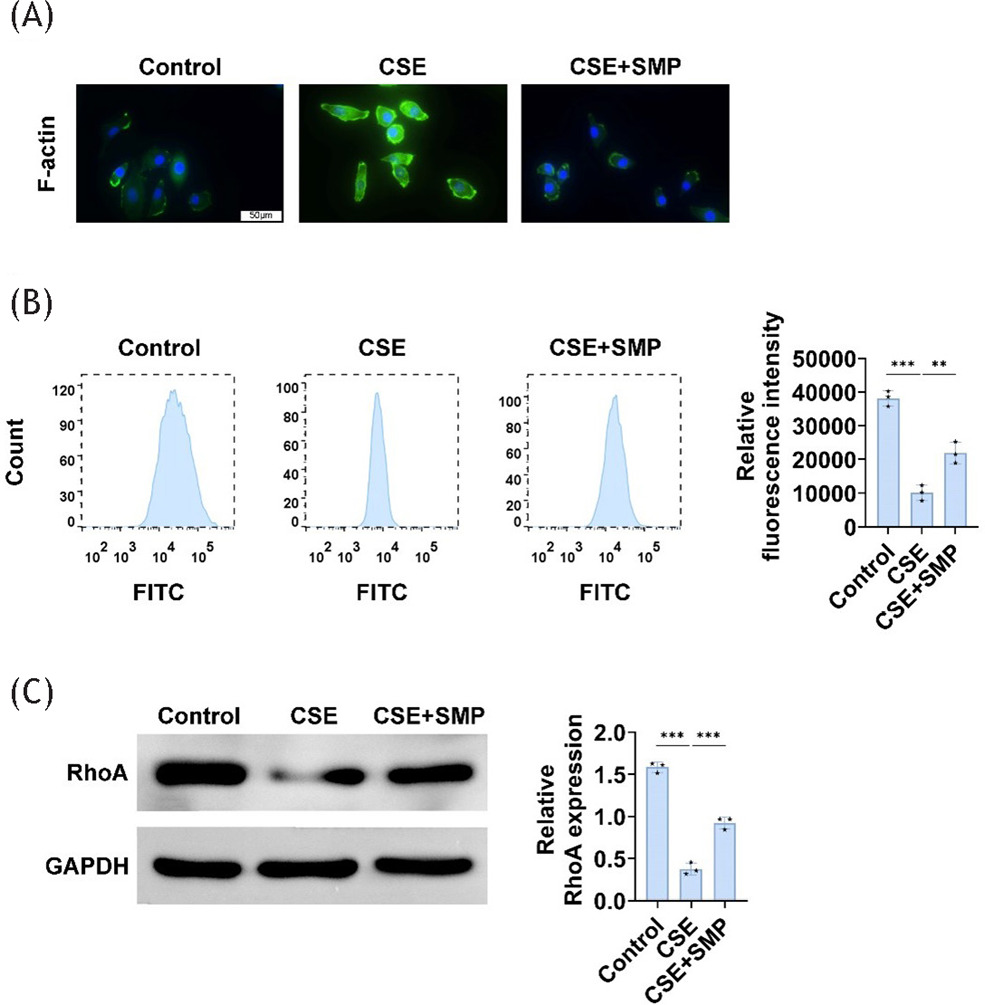
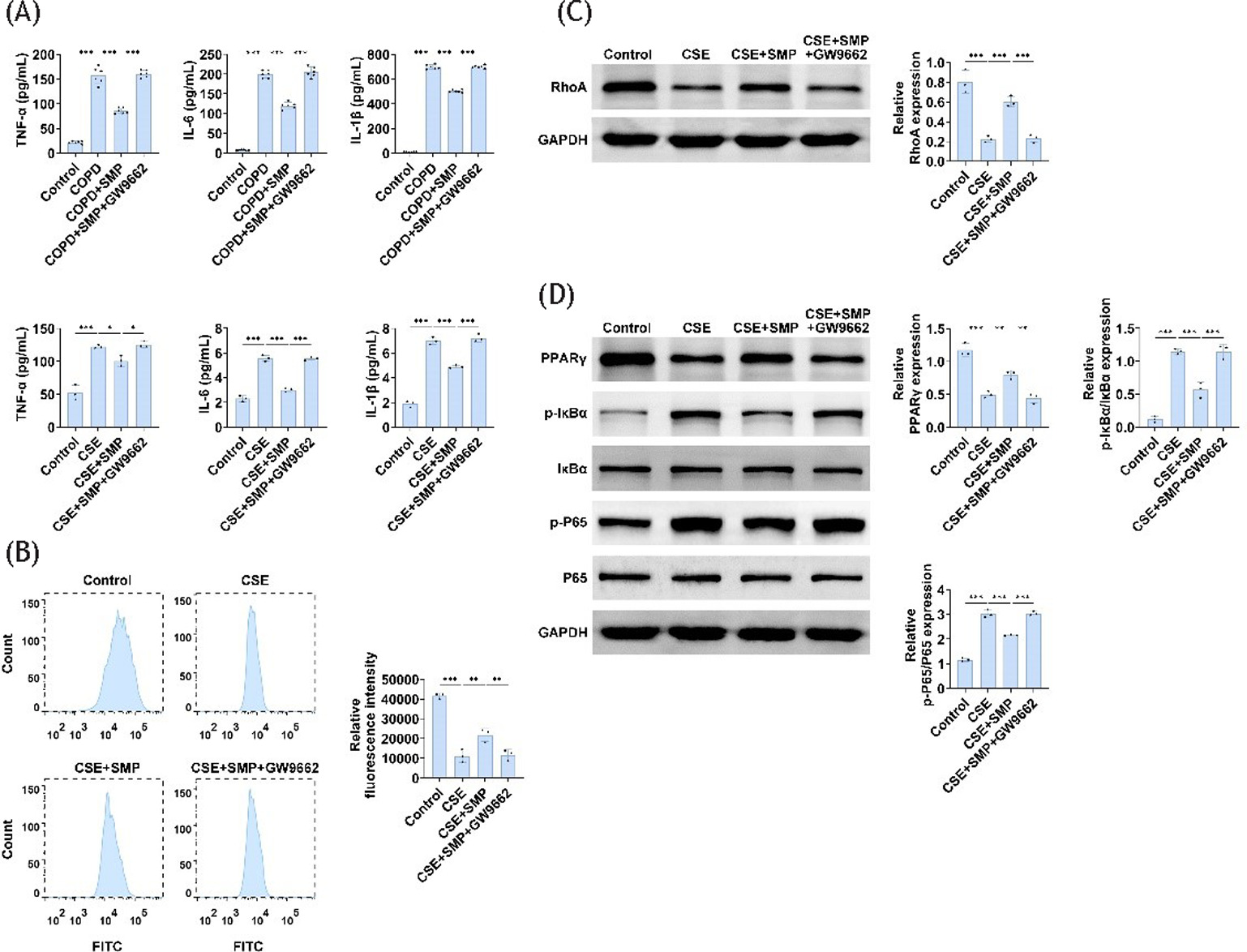
Phagocytosis is essential for macrophage immune defense and tissue remodeling/homeostasis. The phagocytic characteristics of MH-S cells exposed to 5% CSE for 24 h were examined. Compared to the control group, the fluorescence intensity of the F-actin ring of cells from the CSE group increased, while the fluorescence percentage decreased. Additionally, CSE significantly downregulated RhoA expression, while SMP was able to reverse these effects (Figure 3A–C). This demonstrates that SMP enhances AM phagocytosis in in vitro models of COPD.
Figure 3 SMP enhances AM’s phagocytosis function. (A) F-actin ring staining image of AM. (B) Flow cytometry measurements of mean fluorescence intensity. (C) Western blotting to detect RhoA expression in cells. Data are presented as mean ± SD. **P<0.01, *** P < 0.001 versus the control group or CSE group. n=3.
SMP inhibits PPARγ and NF-κB signaling pathway expression
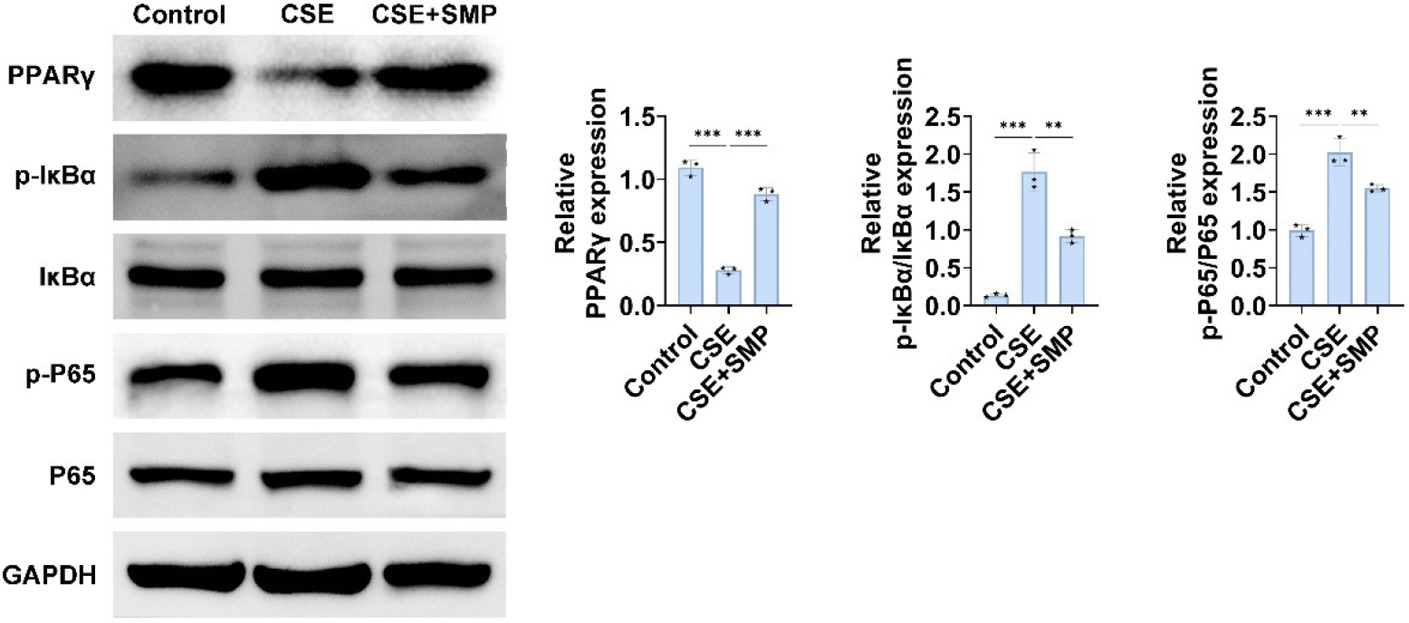
To explore the mechanisms by which SMP treats COPD, we investigated relevant signaling pathways. Compared to the control group, PPARγ expression was decreased, while IκBα and p65 phosphorylation were significantly increased in the CSE group. SMP was able to reverse these effects (Figure 4). These results indicate that SMP may improve COPD through the PPARγ/NF-κB pathway.
Figure 4 SMP inhibits PPARγ and NF-κB signaling pathway expression. Western blotting to detect PPARγ, IκBα, p-IκBα, p65, p-p65 expression in cells. Data are presented as mean ± SD. ** P < 0.01, *** P < 0.001 versus the control group or CSE group. n=3.
SMP improves COPD inflammation and AM’s anti-inflammatory and phagocytic functions by regulating PPARγ
To prove whether SMP improves COPD by regulating PPARγ, we used the PPARγ inhibitor GW9662 for verification. PPARγ inhibition led to a significant increase in inflammatory factors in both lung tissue and AM compared to the SMP-administered group (Figure 5A,B). Additionally, PPARγ inhibition resulted in a significant decrease in RhoA expression and fluorescence percentage in MH-S cells, along with increased phosphorylation levels of IκBα and p65 (Figure 5C,D). These findings indicate that SMP acts through the PPARγ/NF-κB signaling pathway, as the inhibition of PPARγ prevents the effects of SMP.
Figure 5 SMP regulates PPARγ to improve COPD inflammation and AM’s anti-inflammatory and phagocytic functions. (A) ELISA determines TNF-α, IL-6, and IL-1β levels in lung tissue and cells. (B) Flow cytometry measurements of mean fluorescence intensity. (C) Western blotting to detect RhoA expression in cells. (D) Western blotting to detect PPARγ, IκBα, p-IκBα, p65, p-p65 expression in cells. Data are presented as mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001 versus control group, COPD group, and CSE group. n=3.
Discussion
Tobacco smoke, which contains 69 known carcinogenic chemicals, is highly lethal. In around 80% of cases, smoking is a major risk factor for COPD. The harmful particles in smoke can irritate the airways and aggravate COPD.15,16 SMP alleviates COPD airway inflammation both in in vivo and in vitro using cigarette smoke and cigarette extract models. In mice exposed to cigarette smoke, AM phagocytic activity decreased, and pro-inflammatory molecules (IL-6, TNF-α, and IL-1β) were expressed at higher levels. PPARγ expression was reduced, while NF-κB signaling pathway was upregulated. SMP reversed cigarette smoke-induced inflammation, restored AM phagocytosis, suppressed NF-κB signaling, and significantly enhanced PPARγ expression. Moreover, SMP's ability to improve COPD is linked to the PPARγ/NF-κB pathway, as demonstrated by the effects of PPARγ inhibition.
COPD patients generally exhibit much higher systemic inflammation levels compared to control participants. Elevated inflammatory biomarkers, including cytokines, are commonly found in sputum and bronchoalveolar lavage fluid. Additionally, cytokine levels tend to rise with the severity of COPD. It is evident from these studies that inflammation is an important factor in COPD.17,18 Also, AM show significant changes in inflammation, senescence and cell death, tissue proliferation, and repair within the pathophysiology of COPD.19 Activated macrophages, for example, produce reactive oxygen species that damage tissues and increase inflammation by secreting cytokines and other mediators.20 COPD patients often exhibit decreased bacterial phagocytosis in their alveolar macrophages, which may lead to infection, inflammation, or acute exacerbations. Additionally, impaired absorption of apoptotic cells by macrophages can result in prolonged inflammation in COPD patients.21 This study found that SMP effectively suppresses the significant elevation of inflammatory variables (IL-6, TNF-α, and IL-1β) in the lung tissue and AM of mice with COPD induced by cigarette smoke. SMP also suppresses phagocytosis parameters, such as AM fluorescence intensity and RhoA expression. This proves that SMP’s anti-inflammatory effects in COPD are related to its ability to enhance AM phagocytosis.
AM polarization is regulated by the nuclear transcription factor PPARγ, which belongs to the nuclear hormone receptor superfamily.22 PPARγ plays an important role in influencing inflammation, tissue injury, and healing, and it can modulate AM polarization toward M2 both positively and negatively. PPAR-γ agonists decrease inflammatory cell infiltration, decrease airway remodeling, and suppress human airway smooth muscle cell proliferation in COPD.23 PPARγ blocks the activation of AM functional factors, such as NF-ĸB.24 The NF-κB pathway is involved in releasing pro-inflammatory mediators that cause persistent lung inflammation, which contributes to COPD pathophysiology and progression. Therefore, downregulating NF-κB activation can enhance the effectiveness of first-line COPD treatment.25,26 Our findings indicate that SMP promotes PPARγ expression and inhibits NF-κB pathway activation in both in vivo and in vitro models of COPD. In addition, PPARγ inhibitors reverse SMP’s effects on inhibiting inflammatory factors in COPD mice and AM, promoting AM phagocytosis, and reducing NF-κB pathway expression. These results suggest that SMP improves COPD through the modulation of PPARγ.
Conclusions
This study demonstrates that SMP has substantial potential as a therapeutic agent for COPD. SMP's effectiveness is also associated with improving COPD inflammation and increasing AM's phagocytic function through PPARγ activation and NF-κB pathway inhibition. Our findings might suggest that SMP could represent a novel approach to COPD management. Further study is needed to validate these results and explore clinical applications of SMP in COPD treatment.
Funding
This work was supported by the Science and technology project in Lianyungang, Jiangsu Province (Grant No. SF2216).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Data sharing does not apply to this article as no new data were created or analyzed in this study.
Competing interests
The authors state that there are no conflicts of interest to disclose.
Ethics approval
Ethical approval was obtained from the Ethics Committee of Lianyungang TCM Hospital Affiliated with Nanjing University of Chinese Medicine.
Author’s contribution
All authors contributed to the study’s conception and design. Material preparation and the experiments were performed by Dongmei Liu and Zongwei Liu. Data collection and analysis were performed by Xunxun Ma, Shengjie Wang, and Jie Lin. The first draft of the manuscript was written by Xiuyan Shi and Xiaoyong Xu and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Lareau SC, Fahy B, Meek P, Wang A. Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. 2019;199(1):P1–P2. 10.1164/rccm.1991P1
2. Mannino DM, Buist AS. Global burden of COPD: Risk factors, prevalence, and future trends. Lancet. 2007;370(9589):765–73. 10.1016/S0140-6736(07)61380-4
3. Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013. 10.2147/COPD.S82518
4. Rao W, Wang S, Duleba M, Niroula S, Goller K, Xie J, et al. Regenerative metaplastic clones in COPD lung drive inflammation and fibrosis. Cell. 2020;181(4):848–64.e18. 10.1016/j.cell.2020.03.047
5. Park EJ, Park YJ, Lee SJ, Lee K, Yoon C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol Lett. 2019;303:55–66. 10.1016/j.toxlet.2018.12.007
6. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: From mice to humans. Immunology. 2020;160(2):126–38. 10.1111/imm.13154
7. Ghosh B, Gaike AH, Pyasi K, Brashier B, Das VV, Londhe JD, et al. Bacterial load and defective monocyte-derived macrophage bacterial phagocytosis in biomass smoke--related COPD. Eur Respir J. 2019;53(2): 1702273. 10.1183/13993003.02273-2017
8. Cao X, Wang Y, Chen Y, Zhao M, Liang L, Yang M, et al. Advances in traditional Chinese medicine for the treatment of chronic obstructive pulmonary disease. J Ethnopharmacol. 2023;307:116229. 10.1016/j.jep.2023.116229
9. Zhang SY, Yang KL, Long ZY, Li WQ, Huang HY. Use of a systematic pharmacological methodology to explore the mechanism of Shengmai Powder in treating diabetic cardiomyopathy. Med Sci Monit. 2020;26:e919029. 10.12659/MSM.919029
10. Zhang Z, Song Y, Zhang X, Wang S, Jia Z, Wang L, et al. Optimized new Shengmai powder ameliorates myocardial fibrosis in rats with heart failure by inhibition of the MAPK signaling pathway. J Ethnopharmacol. 2024;319(Pt 1):117210. 10.1016/j.jep.2023.117210
11. Wang K, Li L, Wang Y, Fang G, Wei M, Ding H, et al. Effect of Baihe Gujin decoction combined with Shengmai powder on the expression of IL-1beta and IL-1Ra in peripheral blood CD14+ monocytes from patients with pulmonary tuberculosis. Cell Mol Biol (Noisy-le-grand). 2022;68(2):60–3. 10.14715/cmb/2022.68.2.9
12. Li L, Zhang Y, Gong J, Yang G, Zhi S, Ren D, et al. Cpt1a alleviates cigarette smoke-induced chronic obstructive pulmonary disease. Exp Ther Med. 2023;25(1):54. 10.3892/etm.2022.11753
13. Zhang MY, Jiang YX, Yang YC, Liu JY, Huo C, Ji XL, et al. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021;269:119090. 10.1016/j.lfs.2021.119090
14. Hsieh MH, Chen PC, Hsu HY, Liu JC, Ho YS, Lin YJ, et al. Surfactant protein D inhibits lipid-laden foamy macrophages and lung inflammation in chronic obstructive pulmonary disease. Cell Mol Immunol. 2023;20(1):38–50. 10.1038/s41423-022-00946-2
15. Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3(10):733–44. 10.1038/nrc1190
16. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–59. 10.1146/annurev.pathol.4.110807.092145
17. Hacievliyagil SS, Gunen H, Mutlu LC, Karabulut AB, Temel I. Association between cytokines in induced sputum and severity of chronic obstructive pulmonary disease. Respir Med. 2006;100(5):846–54. 10.1016/j.rmed.2005.08.022
18. Karadag F, Karul AB, Cildag O, Yilmaz M, Ozcan H. Biomarkers of systemic inflammation in stable and exacerbation phases of COPD. Lung. 2008;186(6):403–9. 10.1007/s00408-008-9106-6
19. Li Y, Yang Y, Guo T, Weng C, Yang Y, Wang Z, et al. Heme oxygenase-1 determines the cell fate of ferroptotic death of alveolar macrophages in COPD. Front Immunol. 2023;14:1162087. 10.3389/fimmu.2023.1162087
20. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249–56. 10.1111/all.13760
21. Berenson CS, Kruzel RL, Eberhardt E, Dolnick R, Minderman H, Wallace PK, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax. 2014;69(9):811–18. 10.1136/thoraxjnl-2013-203669
22. Furth PA. Peroxisome proliferator-activated receptor gamma and BRCA1. Endocr Relat Cancer. 2019;26(2):R73–R79. 10.1530/ERC-18-0449
23. He S, Tian R, Zhang X, Yao Q, Chen Q, Liu B, et al. PPARγ inhibits small airway remodeling through mediating the polarization homeostasis of alveolar macrophages in COPD. Clin Immunol. 2023;250:109293. 10.1016/j.clim.2023.109293
24. Luo J, Wang J, Zhang J, Sang A, Ye X, Cheng Z, et al. Nrf2 deficiency exacerbated CLP-induced pulmonary injury and inflammation through autophagy-and NF-κB/PPARγ-mediated macrophage polarization. Cells. 2022;11(23): 3927. 10.3390/cells11233927
25. Xu H, Chen X, Liu D, Yang Y. Hispidulin protective impact on sepsis induced acute kidney injury is mediated by regulation of AKT and NF-κB pathway. Signa Vitae. 2023;19(6):152–9. 10.22514/sv.2023.109
26. Yao H, Rahman I. Current concepts on the role of inflammation in COPD and lung cancer. Curr Opin Pharmacol. 2009;9(4):375–83. 10.1016/j.coph.2009.06.009