
Download

ORIGINAL ARTICLE
NLRX1 increases human retinal pigment epithelial autophagy and reduces H2O2-induced oxidative stress and inflammation by suppressing FUNDC1 phosphorylation and NLRP3 activation
Qian Wang, Fengying He*, Liping Wu
Department of Ophthalmology, Huzhou Central Hospital, Huzhou, Zhejiang Province, China
Abstract
Background: Age-related macular degeneration (AMD) is a leading cause of impaired vision as well as some earlier effects, such as reading and face recognition. Oxidative damage and inflammation of retinal pigment epithelial (RPE) cells are major causes of AMD. Additionally, autophagy in RPE cells can lead to cellular homeostasis under oxidative stress. Nucleotide-binding oligomerization domain (NOD)-like receptor X1 (NLRX1) is a mysterious modulator of the immune system function which inhibits inflammatory response, attenuates reactive oxygen species (ROS) production, and regulates autophagy. This study attempted to explore the role of NLRX1 in oxidative stress, inflammation, and autophagy in AMD.
Methods: An in vitro model of AMD was built in human retinal pigment epithelial cell line 19 (ARPE-19) treated with H2O2. The cell viability, NLRX1 expressions, levels of superoxide dismutase (SOD), glutathione (GHS), and ROS, concentrations of interleukin (IL)-1β, tumor necrosis factor-α (TNF-α), IL-6, and monocyte chemoattractant protein-1 (MCP-1), expressions of NLRX1, p62, LC3-II/LC3-I, FUNDC1, and NOD-like receptor protein 3 (NLRP3) inflammasome were expounded by cell counting kit-8, colorimetric, enzyme-linked immunosorbent serologic assay (ELISA), and Western blot assay.
Results: H2O2 treatment notably reduced the relative protein expression of NLRX1. Meanwhile, H2O2 incubation decreased cell viability, diminished SOD and GSH concentrations, accompanied with the increased level of ROS, enhanced IL-1β, TNF-α, IL-6, and MCP-1 concentrations, and aggrandized the relative protein expression of p62 with reduced LC3-II/LC3-I ratio. Moreover, these results were further promoted with knockdown of NLRX1 and reversed with overexpression. Mechanically, silencing of NLRX1 further observably enhanced the relative levels of -phosphorylated FUNDC1/FUNDC1, and NLRP3 inflammasome-related proteins, while overexpression of NLRX1 exhibited inverse results in the H2O2-induced ARPE-19 cells.
Conclusion: NLRX1 suppressed H2O2-induced oxidative stress and inflammation, and facilitated autophagy by suppressing FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells.
Key words: age-related macular degeneration, autophagy, FUNDC1, Inflammation, NLRX1, NLRP3 inflammasome, oxidative stress
*Corresponding author: Fengying He, Department of Ophthalmology, Huzhou Central Hospital, No. 1558 North Sanhuan Road, Huzhou, Zhejiang Province 313000, China. Email address: [email protected]
Received 30 August 2022; Accepted 4 November 2022; Available online 1 January 2023
Copyright: Wang Q, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Age-related macular degeneration (AMD) is a prevalent retinal eye disease which is characterized by injury to the macular region of the retina involved in central vision acuity,1 and appeared as choroidal neovascularization (CNV) or geographic atrophy and drusen in the macula.2,3 The manifestations of AMD are inability to see fine details and colors clearly, and blurring or dark spots in the middle part of the visual field, thereby resulting in progressive loss of central vision. AMD is a leading cause of blindness among the elderly, with a prevalence of 8.69% in people aged 45–85 years in 2014.4 Moreover, it has been estimated that AMD because of its steadily increasing rate would affect 288 million people globally by 2040.5 The pathogenesis of AMD is complicated and remains to be explored. Nevertheless, several molecular mechanisms, such as inflammation, oxidative stress, autophagy, and impaired angiogenesis, have been expounded to be strongly associated with the process of AMD.2,6 Numerous treatments, including photodynamic therapy and anti-vascular endothelial growth factor (VEGF) injections, have been introduced for managing AMD. However, they severely lack the approaches to protect light-sensitive cells in the macula from progressive irreversible degeneration, particularly in the atrophic AMD.3 Therefore, in-depth exploration of the molecular mechanism of AMD and screening of potential therapeutic targets are crucial for the clinical treatment of AMD.
The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family covers evolutionarily conserved constituents of the immune system and exercises crucial roles in inflammation and immune defense.7 NOD-like receptor X1 (NLRX1) is a unique member of NLR family that negatively modulates the expression level of pro-inflammatory factors and serves the role of inflammasome.8,9 In addition, NLRX1 has been demonstrated to participate in diverse pathological progresses because of its wide variety of biological functions, such as apoptosis, autophagy, modulation of mitochondrial injury, regulation of reactive oxygen species (ROS) generation, negative modulation of signaling pathways, including nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK), c-Jun N-terminal Kinase (JNK), and interferon-1 (IFN-1).10,11 Thus, NLRX1 has been involved in multitudinous diseases, including the nervous, circulatory, respiratory, urinary, motor, and digestive systems.12 Additionally, NLRX1 can regulate another key factor in mitophagy, FUN14 domain containing 1 (FUNDC1), in which it can activate FUNDC1 through dephosphorylation of Tyr 18 of FUNDC1, induce autophagy, and relieve intestinal oxidative stress and inflammation caused by ischemia-reperfusion.13 FUNDC1 is a mammalian mitochondrial autophagy receptor expressed in outer membrane of the mitochondria.14 FUNDC1 has been underlined to activate autophagy via directly binding to LC3,15,16 and regulates hypoxia-induced mitochondrial autophagy.17 Furthermore, FUNDC1 is able to regulate autophagy by inhibiting ROS–NOD-like receptor protein 3 (NLRP3) signaling to avoid lipopolysaccharide-induced lung apoptosis in mouse models.18 Nevertheless, the role and underlying molecular mechanisms of NLRX1 in AMD require more analyses.
In the current study, an in vitro model of AMD was built in human retinal pigment epithelial cell line 19 (ARPE-19) treated with H2O2. The role and mechanisms of NLRX1 were revealed in H2O2-induced ARPE-19 cell line. We hope our results could lay a theoretical basis for the clinical diagnosis and treatment of AMD.
Materials and methods
Cell culture
Human retinal epithelial cells 19 (ARPE-19, CL-0026; Procell, Wuhan, China) were maintained in Dulbecco's modified eagle medium (DMEM)/nutrient mixture F-12 (PM150312; Procell) provided with 10% fetal bovine serum (FBS, 164210-50; Procell) and 1% penicillin/streptomycin (PB180120; Procell) in an incubator with 5% carbon dioxide (CO2) at 37°C. All ARPE-19 cells used in this study were third to seventh passages after recovery.
Cell treatment and transfection
ARPE-19 cells were challenged with 300-μM H2O2 (diluted into phosphate buffer saline (PBS, P1020; Solarbio, Beijing, China) for 24 h to induce an in vitro model of AMD, as done in a previous study.19 To knockdown of expression of NLRX1, small interfering (si)RNAs targeting NLRX1 (siNLRX1) and the relevant negative control (siNC) were acquired from GenePharma (Shanghai, China). On the other hand, plasmid cloning (pc)DNA vector plasmids hiding the sequences of NLRX1 or the empty pcDNA vector plasmids (vector) were obtained to overexpress the level of NLRX1. All the siNLRX1, siNC, NLRX1, and vector were transfected into ARPE-19 cells with lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Cells were harvested through centrifugation with 1000 g for 5 min after transfection for 48 h and treated with 300-μM H2O2 for 24 h.
Cell counting kit-8 (CCK-8) analysis
The viability of ARPE-19 cells was examined by CCK-8 cell proliferation and cytotoxicity assay kit (CA1210; Solarbio) as described in a previous paper.20 In brief, following H2O2 treatment with or without transfection, ARPE-19 cells were seeded into 96-well plates with 40,000 cells/well, and maintained with 5% CO2 at 37°C for 24 h. Then, 10-µL of CCK-8 reagents were added into each well for further incubation of 2 h. The optical density (OD) was tested using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm. Data were yielded from at least 10,000 cells.
Western blot analysis
Based on previous studies,21,22 total proteins from cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (R0010; Solarbio), harvested through centrifugation with 12,500 g for 5 min at 4°C, and quantified with the bicinchoninic acid (BCA) protein assay kit (PC0020; Solarbio) according to operating instructions. Protein samples, 20 µg, were dissolved with sodium dodecyl sulfate–-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrically onto a polyvinylidene fluoride (PVDF) membrane (EMD Millipore; Billerica, MA, USA). Then, 3% bovine serum albumin (BSA) was used to seal the membranes for 60 min at room temperature. Next, the membranes were treated with primary antibodies overnight at 4°C. The second antibody goat anti-rabbit immunoglobulin G (IgG) H&L (HRP) (1:20,000; Abcam, Cambridge, UK) was hatched with the membranes at room temperature for 2 h. Subsequently, the membranes were visualized by the ECL Western blotting substrate (PE0010; Solarbio). The gray value was quantified by QUANTITY ONE software (Bio-Rad, Hercules, CA, USA). The primary antibodies included rabbit anti-NLRX1 (1:1000, ab107611; Abcam), rabbit anti-p62 (1:1000, ab91526; Abcam), rabbit anti-LC3-I/II (1:1000, ab128025; Abcam), rabbit anti-FUNDC1 (1:5000, ab224722; Abcam), rabbit anti-NLRP3 (1:1000, 15101; Cell Signaling Technology Inc., Danvers, MA, USA), rabbit anti-apoptosis-associated speck-like protein (ASC) containing a caspase recruitment domain (CARD) (1:2000, ab180799; Abcam), rabbit anti--pro-caspase 1 (1:1000, 2225; Cell Signaling Technology Inc., Danvers, MA, USA), rabbit anti-cleaved-caspase 1 (1:1000, 4199; Cell Signaling Technology), and rabbit anti--glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:2500, ab9485; Abcam). The primary antibodies of phosphorylated (p-)FUNDC1 (Tyr18; 1:500) were generated through immunizing rabbits with synthesized and purified phosphorylated peptides from FUNDC1 (Abgent, Suzhou, China) based on previous studies.13,23
Measurement of superoxide dismutase (SOD) and glutathione (GSH) levels
The concentrations of SOD and GSH were detected with the SOD assay kit (A001-3-2) and the total GSH/oxidized GSH assay kit (A061-2-1; both from Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to operating directions at 450 nm and 405 nm with a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA), separately. Data were yielded from at least 10,000 cells.
Detection of ROS level
ARPE-19 cells with a density of 2.5 × 105 cells/well were plated into 24-well plates and maintained at 37°C for 48 h. Following H2O2 induction and transfection, cells were -collected and rinsed and then administrated with 10-µM 5-(and-6)-chloromethyl-2-,7-dichlorofluorescin diacetate (DCFH-DA, D6470; Solarbio) in dark at 37°C for 30 min. The images were obtained by a fluorescence microscope (Olympus, Tokyo, Japan). Data were yielded from at least 10,000 cells.
Enzyme-linked immunosorbent serologic assay (ELISA)
The cell culture media were centrifuged at 1000×g to remove debris. Then, the concentrations of interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and monocyte chemoattractant protein (MCP)-1 in cell culture supernatant were measured by human IL-1β ELISA kit (SEKH-0002; Solarbio), Human TNF-α ELISA kit (SEKH-0047; Solarbio), human MCP-1 ELISA kit (SEKH-0236; Solarbio), and Human IL-6 ELISA kit (SEKH-0013; Solarbio) according to the manufacturer’s protocol.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was obtained from ARPE-19 cells by TRIzol reagent (15596026; Thermo Fisher). Total RNA, 1 μg, was reversely transcribed into complementary DNA (cDNA) by a PrimeScript RT reagent kit (RR047A; Takara, Dalian, China). RT-qPCR was carried out by using TB Green TM Premix Ex TaqTM II (Tli RNaseH Plus) (RR820A; Takara) on the A PIKORed 96 (ThermoFisher). The PCR amplification conditions were 95°C for 10 min, 95°C for 10 s, and 56°C for 30 s for 40 cycles. GAPDH served as an internal reference. The expressions of genes were analyzed with the comparative threshold cycle method (2-ΔΔCT method), in which ΔΔCT = ΔCTtreatment - ΔCTcontrol and ΔCT = Cttarget - Ctreference. Primer sequences were NLRP3: 5'-GCACCTGTTGTGCAATCTGAA-3' (forward) and 5'-TCCTGACAACATGCTGATGTGA-3' (reverse), and GAPDH: 5'- TGCACCACCAACTGCTTCGC-3' (forward) and 5'-GGCATGGACTGTGGTCATGAG-3' (reverse).
Statistical analysis
The experiments were performed with three replicates, and the results were expressed as mean ± standard deviation (SD). The one-way analysis of variance (ANOVA) was used for statistical analysis of the data, followed by post hoc Bonferroni test by the SPSS 26.0 software (IBM, Armonk, NY, USA); P ˂ 0.05 was considered as statistically significant.
Results
H2O2 induced the downregulation of NLRX1 in ARPE-19 cells
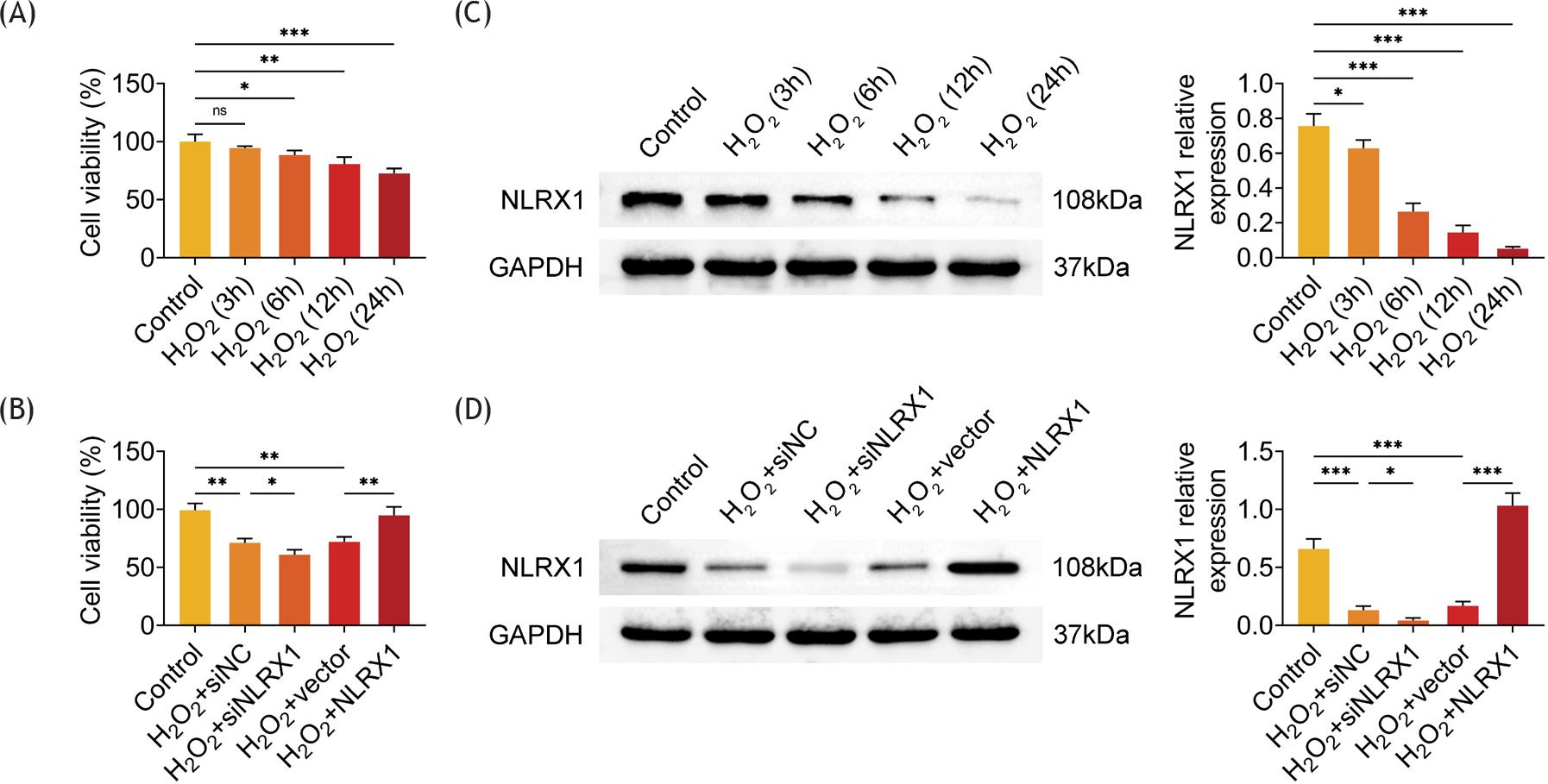
In order to investigate the role of NLRX1 in ARPE-19 cells, cells were treated with 300-μM H2O2 for 3, 6, 12, and 24 h. H2O2 induced a prominent decrease in the viability of ARPE-19 cells, compared to that in cells without H2O2 treatment (control group) in a time-dependent manner (Figure 1A). Meanwhile, H2O2 administration also significantly downregulated the relative protein expression of NLRX1 in ARPE-19 cells, compared to the control group in a time-dependent manner (Figure 1B). Compared to the cells in the control group without transfection and H2O2 treatment, the cell viability notably diminished in H2O2-induced ARPE-19 cells transfected with siNC (Figure 1C). Moreover, transfection of siNLRX1 into H2O2-induced ARPE-19 cells further prominently reduced cell viability, while overexpression of NLRX1 in H2O2-induced ARPE-19 cells notably rescued cell viability, compared to that in H2O2-induced cells transfected with siNC (Figure 1C). Correspondingly, silencing of NLRX1 further observably diminished the relative protein expression of NLRX1 whereas upregulation of NLRX1 markedly restored the relative protein expression of NLRX1 in H2O2-treated ARPE-19 cells (Figure 1D). Thus, H2O2 induced downregulation of NLRX1 and reduction of viability in ARPE-19 cells.
Figure 1 Analysis of viability and expression of NLRX1 in ARPE-19 cells. (A) The cell viability was determined by CCK-8 assays after ARPE-19 cells were treated with 300-μM H2O2 for 0, 3, 6, 12, and 24 h. (B) The relative protein expression of NLRX1 in ARPE-19 cells treated with 300-μM H2O2 for 0, 3, 6, 12, and 24 h was determined by Western blot analysis. The data were expressed after normalized with GAPDH. (C) The cell viability was measured by CCK-8 assays after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids, and their corresponding negative control (NC). After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. (D) The relative protein expression of NLRX1 was examined by Western blot analysis after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids, and their corresponding negative control. After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. The data were expressed after normalized with GAPDH. The experiments were performed with three replicates, and the data were expressed as mean ± SD. *P ˂ 0.05, **P ˂ 0.01, and ***P ˂ 0.001; ns: nonsignificant.
NLRX1 inhibited oxidative stress in H2O2-evoked ARPE-19 cells
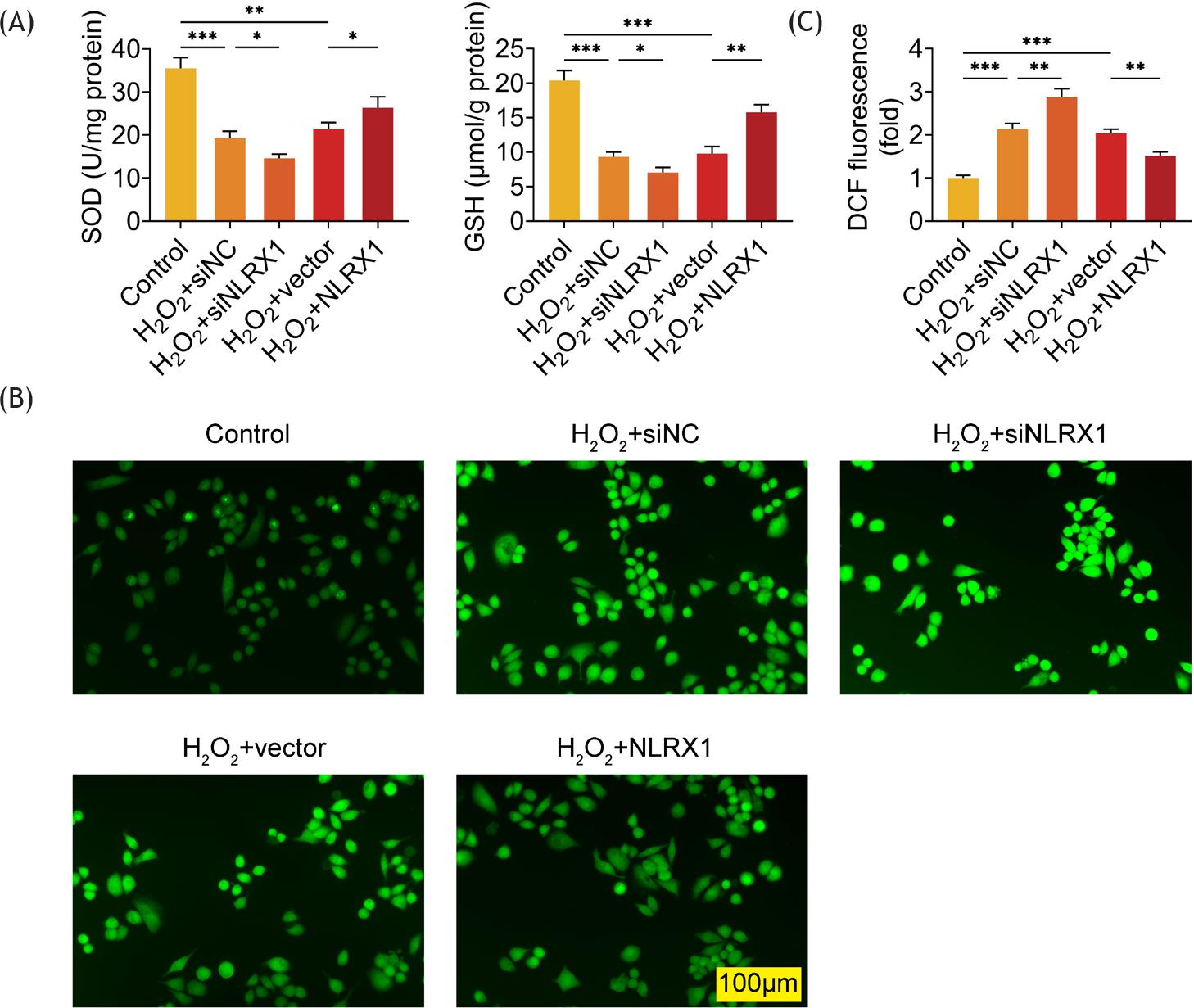
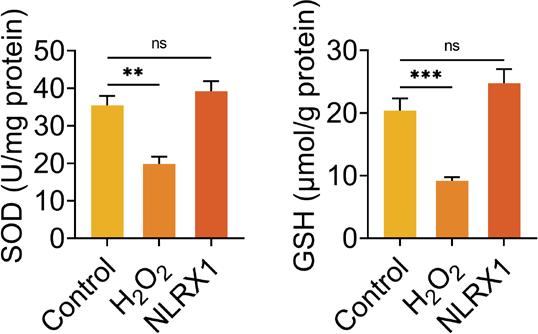
Then, the role of NLRX1 in oxidative stress was discussed in ARPE-19 cells through the detection of SOD, GSH, and ROS levels. H2O2 treatment had no influence on the concentrations of SOD and GSH, while overexpression of NLRX1 markedly increased the concentrations of SOD and GSH in ARPE-19 cells (Supplementary Figure S1). Compared to the cells in the control group without transfection and H2O2 treatment, the SOD and GSH concentrations observably diminished in H2O2-induced ARPE-19 cells transfected with siNC (Figure 2A). Interference of NLRX1 further notably reduced the concentrations of SOD and GSH in H2O2-treated ARPE-19 cells, while overexpression of NLRX1 lead to a reverse result in the concentrations of SOD and GSH in H2O2-treated ARPE-19 cells, compared to those in the H2O2+siNC group (Figure 2A). On the contrary, knockdown of NLRX1 further significantly boosted the level of ROS, and upregulation of NLRX1 markedly neutralized the level of ROS in H2O2-treated ARPE-19 cells (Figures 2B and C). Therefore, NLRX1 suppressed oxidative stress in H2O2-induced ARPE-19 cells.
Figure 2 Examination of the levels of SOD, GSH and ROS. (A) The concentrations of SOD and GSH were measured through commercial kits, after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids and their corresponding NC. After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. (B and C) ARPE-19 cells were transfected with siNLRX1, overexpression plasmids and their corresponding NC. After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h, and then the level of ROS was imaged by fluorescence microscope after ARPE-19 cells were loaded with DCFH-DA. Scale bar = 100 μm for all images. The experiments were performed with three replicates, and the data were expressed as mean ± SD. *P ˂ 0.05, **P ˂ 0.01, and ***P ˂ 0.001.
NLRX1 restrained inflammation in H2O2-induced ARPE-19 cells
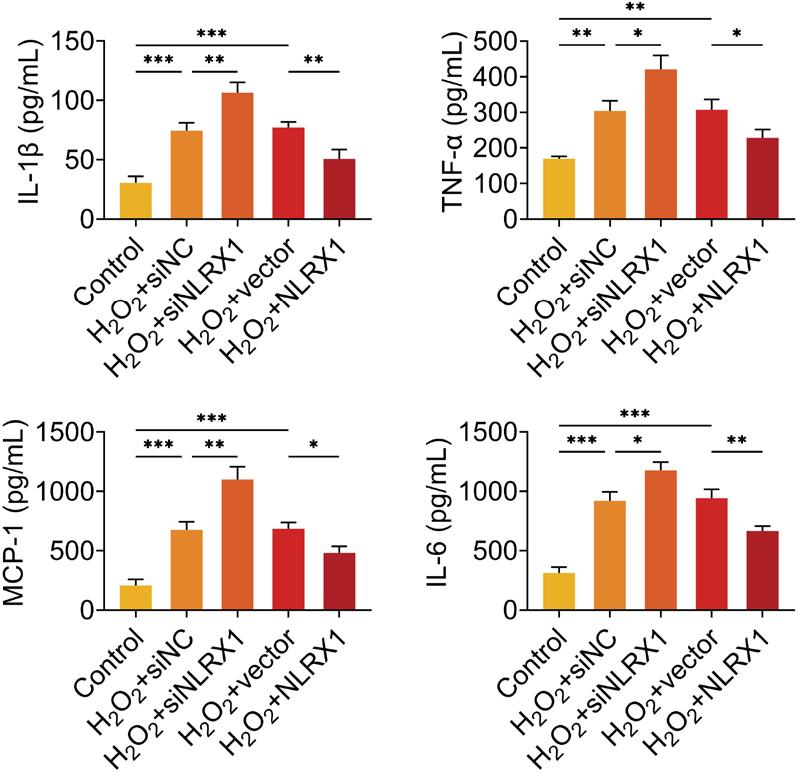
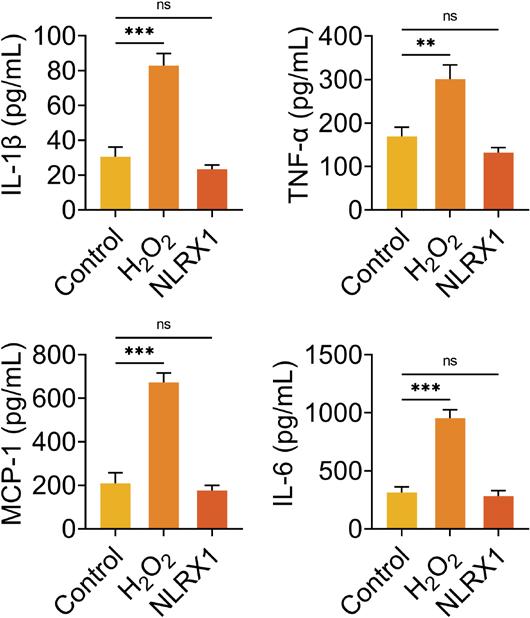
The levels of cytokine release were detected after ARPE-19 cells were treated with H2O2 alone or overexpressed with NLRX1. The results demonstrated that H2O2 treatment alone observably enhanced the concentrations of IL-1β, TNF-β, MCP-1, and IL-6, while overexpression of NLRX1 had no influence on the concentrations of IL-1β, TNF-β, MCP-1, and IL-6 in ARPE-19 cells (Supplementary Figure S2). Results demonstrated in Figure 3 revealed that compared to the cells in the control group without transfection and H2O2 treatment, the concentrations of L-1β, TNF-β, MCP-1, and IL-6 were observably elevated in H2O2-induced ARPE-19 cells transfected with siNC (Figure 3). Moreover, the concentrations of IL-1β, TNF-β, MCP-1, and IL-6 were dramatically augmented with the knockdown of NLRX1, and signally declined with the upregulation of NLRX1 H2O2-induced ARPE-19 cells, compared to those in the H2O2+siNC group (Figure 3). Hence, NLRX1 repressed H2O2-induced inflammation in ARPE-19 cells.
Figure 3 Measurement of the concentrations of IL-1β, TNF-α, MCP-1 and IL-6. The concentrations of IL-1β, TNF-α, MCP-1 and IL-6 were detected with commercial kits, after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids and their corresponding NC. After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. The experiments were performed with three replicates, and the data were expressed as mean ± SD. *P ˂ 0.05, **P ˂ 0.01, and ***P ˂ 0.001.
NLRX1 restored H2O2-blocked autophagy in ARPE-19 cells
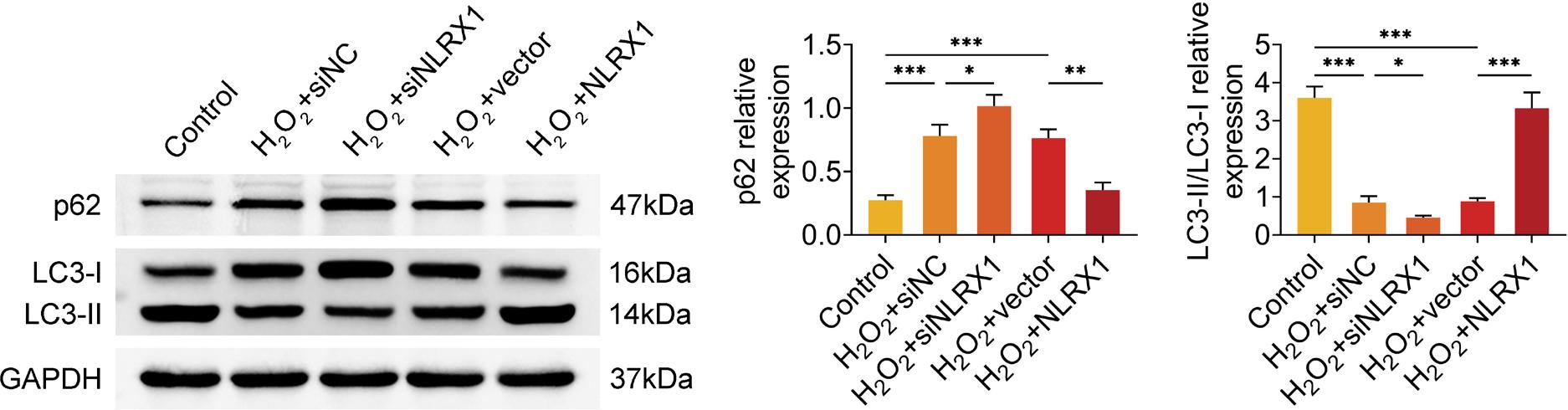
As demonstrated in Figure 4, the relative protein expression of p62 was significantly upregulated and the LC3II/LC3I ratio was prominently downregulated in H2O2-induced ARPE-19 cells transfected with siNC, compared to the cells in the control group without transfection and H2O2 treatment. Moreover, an outstanding increase in the p62 protein levels and a remarkable decrease in the LC3II/LC3I ratio were indicated in H2O2-induced ARPE-19 cells transfected with siNLRX1, compared to those in the H2O2+siNC group (Figure 4). Meanwhile, inverse results were observed after H2O2-induced ARPE-19 cells overexpressed with NLRX1 (Figure 4). Hence, H2O2 inhibited autophagy, which was rescued by NLRX1 in ARPE-19 cells.
Figure 4 NLRX1 enhanced H2O2-induced autophagy in ARPE-19 cells. The relative protein expressions of p62, LC3-II, and LC3-I were detected by western blot, after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids and their corresponding negative control (NC). After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. The data were expressed after normalized with GAPDH. The experiments were performed with three replicates, and the data were expressed as mean ± SD. *P ˂ 0.05, **P ˂ 0.01, and ***P ˂ 0.001.
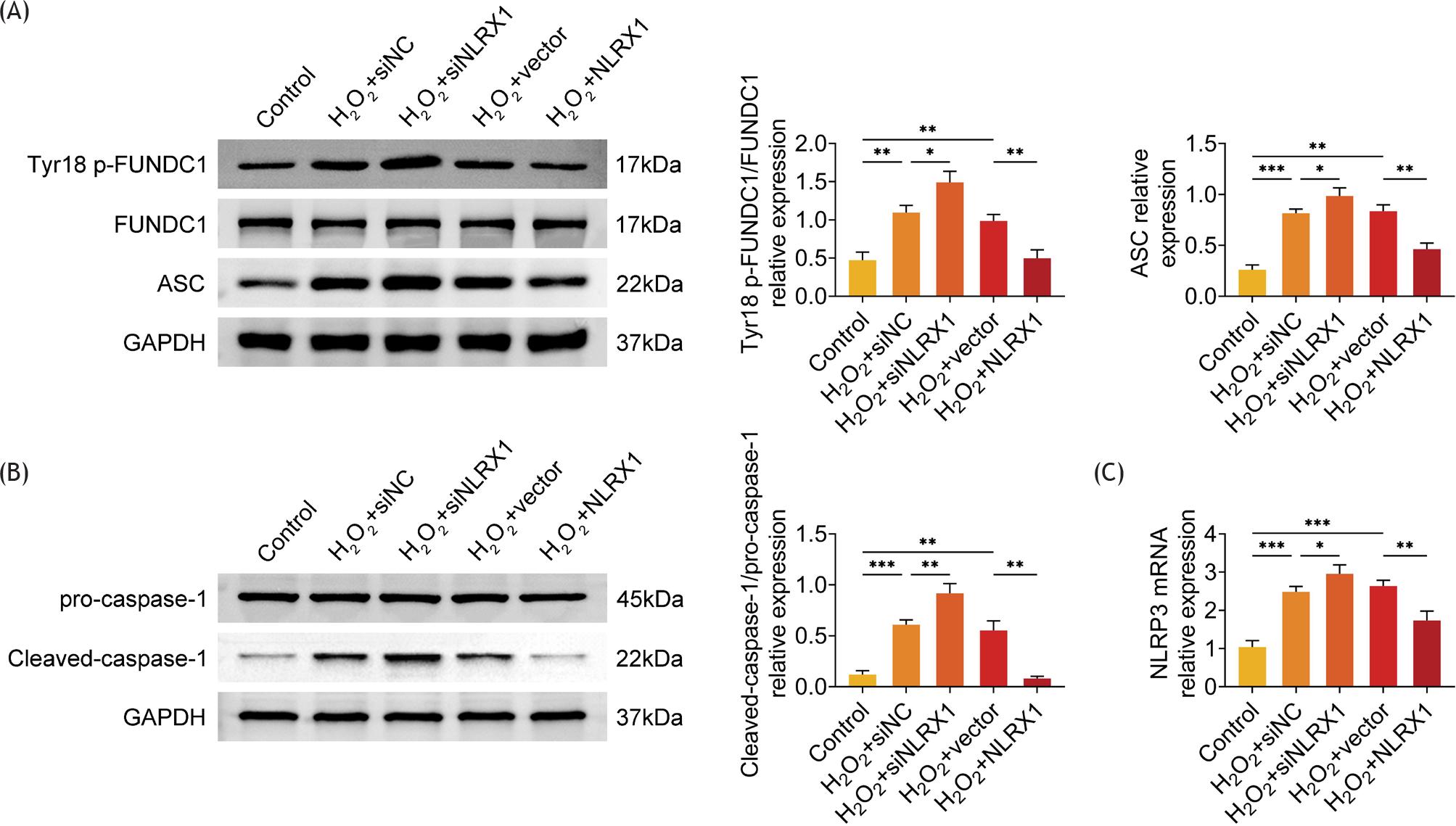
NLRX1 suppressed FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells
Furthermore, the possible molecular mechanism was investigated by examining the relative protein expressions of FUNDC1, p-FUNDC1 (Tyr 18), and NLRP3 inflammasome-related proteins. The results revealed that silencing of NLRX1 further observably enhanced the relative protein levels of p-FUNDC1/FUNDC1, while overexpression of NLRX1 prominently reduced the relative protein levels of p-FUNDC1/FUNDC1 in H2O2-induced ARPE-19 cells (Figure 5A). Since the protein level of NLRP3 detected by Western blot analysis was extremely low (Figure S3), the protein expression of caspase-1 and the messenger RNA (mRNA) level of NLRP3 were examined in H2O2-treated ARPE-19 cells. As shown in Figures 5B and C, NLRX1 inhibited the relative protein level of cleaved-caspase-1/pro-caspase-1 and the transcriptional expression of NLRP3 in H2O2-induced ARPE-19 cells. Therefore, NLRX1 refrained FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells.
Figure 5 NLRX1 inhibited FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells. (A and B) The relative protein expression of FUNDC1, phosphorylated FUNDC1 (Tyr 18) and ASC, pro-caspase-1, and cleaved-caspased-1 were determined by Western blot analysis, after ARPE-19 cells were transfected with siNLRX1, overexpression plasmids and their corresponding negative control (NC). After transfection for 48 h, cells were treated with 300-μM H2O2 for 24 h. The data were expressed after normalized with GAPDH. (C) The relative mRNA level of NLRP3 was measured by RT-qPCR. The data were expressed after normalized with GAPDH. The experiments were performed with three replicates, and the data were expressed as mean ± SD. *P ˂ 0.05, **P ˂ 0.01, and ***P ˂ 0.001.
Discussion
In the present study, an in vitro model of AMD was built in ARPE-19 cells treated with H2O2. NLRX1 suppressed H2O2-induced oxidative stress and inflammation, and restored H2O2-blocked autophagy by inhibiting the FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells.
NLRX1 is a negative modulatory NLR that plays a suppressive role in inflammatory signaling.8,9 It has been revealed to be dysregulated in various disease models. NLRX1 has been discovered to be downregulated in celiac disease though transcriptomics data,24 and intestinal ischaemic reperfusion injury.13 Moreover, Pickering et al.25 summarized the emerging roles of NLRX1 in human diseases, in which NLRX1 was strongly associated with neuroinflammatory disorders, inflammatory bowel disease, autoimmunity, metabolic disease, ischaemia reperfusion injury, and cancers. In the mentioned study, the relative protein level of NLRX1 was also downregulated in the H2O2-induced ARPE-19 cells with a prominent decrease in the viability of ARPE-19 cells, which was confirmed with the loss-of-function and gain-of-function assays.25 Thus, downregulation of NLRX1 was observed in the H2O2-treated ARPE-19 cells and was closely involved in cell viability.
Inflammation, oxidative stress, and autophagy are pivotal pathogeneses involved in a variety of diseases.26–29 Similarly, the above pathogeneses are also tightly involved with AMD progression. Inflammation exerts a vital role in the etiology and pathogenesis of AMD, thus a series of therapeutic approaches that target the reduction in chronic inflammation have been applied to postpone or prevent retinal degeneration in AMD.30,31 Also, oxidative stress is a mechanism related to the pathogenesis and progression of AMD, in which oxidative stress-induced retinal pigment epithelial (RPE) injury is one of the dominating causes. Pentraxin 3 induced by oxidative stress promotes RPE cell death correlated to the development of AMD.32 Oxidative stress decreased the ability of RPE stem cells and influenced the onset and progression of AMD.33 In addition, Mitter et al. reported that impairment of autophagy promoted the progression of AMD.34 Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) was reported to be a putative therapy targeted against AMD, which was involved in oxidative stress and autophagy.35 Consistently, our results have revealed that H2O2 treatment promoted oxidative stress and inflammation, and inhibited autophagy in ARPE-19 cells. Moreover, NLRX1 inhibited inflammatory responses in chondrocytes36 and myocardial ischemia8 NLRX1 suppressed inflammation and oxidative stress in inflammatory bowel disease both in vitro and in vivo.37 Aikawa et al. demonstrated that NLRX1 negatively modulated invasion and autophagy through interaction with the Beclin 1–ultraviolet radiation resistance-associated gene protein (UVRAG) complex during Group A streptococcus infection.38 Thus, these findings indicated that NLRX1 mediated inflammation, oxidative stress, and autophagy in different disease models. Here, the results also revealed that NLRX1 inhibited inflammation and oxidative stress with enhanced autophagy in the H2O2-induced ARPE-19 cells via both loss-of-function and gain-of-function assays. Altogether, NLRX1 inhibited H2O2-induced oxidative stress and inflammation, and restored H2O2-blocked autophagy in ARPE-19 cells.
Mechanically, NLRX1 repressed FUNDC1 phosphorylation and NLRP3 activation in H2O2-induced ARPE-19 cells. Li et al. demonstrated that NLRX1 activated FUNDC1 through dephosphorylation of Tyr 18, which mediated induced inflammation, oxidative stress, and mitophagy in intestinal ischaemic reperfusion injury.13 Moreover, FUNDC1 modulated autophagy by suppressing ROS–NLRP3 signaling, which prevented the lung from apoptosis in a lipopolysaccharide (LPS)-induced mouse model.18 NLRP3 inflammasome grouped with the adaptor, that is, apoptosis-associated speck-like protein (ASC) containing CARD that enhanced the binding of the pyrin domain (PYD) at the N-terminus of NLRP3 and the CARD domain of the effector pro-caspase-1 to recruit NLRP3.39 NLRP3 inflammasome effectively promoted the modulation of immune responses and the recruitment of inflammatory cells in various organs.40 Therefore, NLRX1 suppressed FUNDC1 phosphorylation and NLRP3 activation in ARPE-19 cells.
Conclusion
The expression of NLRX1 was downregulated in H2O2-induced ARPE-19 cells. Results of both loss-of-function and gain-of-function assays revealed that NLRX1 suppressed H2O2-induced oxidative stress and inflammation, and facilitated autophagy, which could be strongly involved in the regulation of FUNDC1 and NLRP3 inflammasome in ARPE-19 cells. Nevertheless, several limitations remained in this study that could be dealt with in the following study. First, an in vivo validation is essential in the future study. Next, direct role between FUNDC1–NLRP3 inflammasome and NLRX1 function must be confirmed through pharmacological block or other effective interventions. In addition, NLRX1, as a mitochondrial protein, had a crucial role in the intestinal ischaemic reperfusion injury by the modulation of mitophagy. Thus, the future study on the role of NLRX1 in the mitophagy of H2O2-induced ARPE-19 cells could be an interesting direction. In addition, dysregulated autophagy contributed to the deterioration of oxidative stress and the pathogenesis of AMD.34 Chronic oxidative stress has been found to induce and/or maintain a pro-inflammatory retinal microenvironment, thus affecting the onset and progression of AMD.33 Hence, the modulatory role of oxidative stress, inflammation, and autophagy must be focused in the prospective study. Briefly, our study indicated that NLRX1 is a potential target for the treatment of AMD.
Conflict of interests
The authors stated that there were no conflicts of interest to disclose.
Ethics approval
This study did not contain experiments with human participants or animals performed by any of the authors.
Data availability
The authors declare that all data supporting the findings of this study are available in the paper, and any raw data can be obtained from the corresponding author upon request.
Author Contributions
Qian Wang and Fengying He designed and performed the study. Qian Wang, Fengying He, and Liping Wu supervised data collection, and analyzed and interpreted the collected data. All the authors prepared and reviewed the draft of the manuscript. All authors read and approved the final manuscript for publication.
REFERENCES
1. Arruabarrena C, Toro MD, Onen M, Malyugin BE, Rejdak R, Tognetto D, et al. Impact on visual acuity in neovascular age related macular degeneration (nAMD) in Europe due to COVID-19 pandemic lockdown. J Clin Med. 2021;10(15):. 10.3390/jcm10153281.
2. Fleckenstein M, Keenan TDL, Guymer RH, Chakravarthy U, Schmitz-Valckenberg S, Klaver CC, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31. 10.1038/s41572-021-00265-2.
3. Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392(10153):1147–59. 10.1016/s0140-6736(18)31550-2.
4. GBD 2019 Blindness and Vision Impairment Collaborators; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. Lancet Glob Health. 2021;9(2):e144–60. 10.1016/s2214-109x(20)30489-7.
5. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–16. 10.1016/s2214-109x(13)70145-1.
6. Zhang ZY, Bao XL, Cong YY, Fan B, Li GY. Autophagy in age--related macular degeneration: A regulatory mechanism of oxidative stress. Oxid Med Cell Longev. 2020;2020:2896036. 10.1155/2020/2896036.
7. Kourelis J, Adachi H. Activation and regulation of NLR immune receptor networks. Plant Cell Physiol. 2022. Epub ahead of print. 10.1093/pcp/pcac116.
8. Li H, Zhang S, Li F, Qin L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol Immunol.2016;76:90–7. 10.1016/j.molimm.2016.06.013.
9. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. immunity. 2011;34(6):843–53. 10.1016/j.immuni.2011.02.022. PMid: 21703539.
10. Nagai-Singer MA, Morrison HA, Allen IC. NLRX1 is a multifaceted and enigmatic regulator of immune system function. Front Immunol. 2019;10:2419. 10.3389/fimmu.2019.02419.
11. Leber A, Hontecillas R, Tubau-Juni N, Zoccoli-Rodriguez V, Abedi V, Bassaganya-Riera J. NLRX1 Modulates Immunometabolic Mechanisms Controlling the Host-Gut Microbiota Interactions during Inflammatory Bowel Disease. Front Immunol. 2018; 9:363. 10.3389/fimmu.2018.00363.PMid: 29535731.
12. Shi H, Zhou ZM, Zhu L, Chen L, Jiang ZL, Wu XT. Underlying Mechanisms and Related Diseases Behind the Complex Regulatory Role of NOD-Like Receptor X1. DNA Cell Biol. 2022; 41(5):469-478. 10.1089/dna.2022.0051. PMid: 35363060.
13. Li S, Zhou Y, Gu X, Zhang X, Jia Z. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021;54(3):e12986. 10.1111/cpr.12986.
14. Lim Y, Rubio-Peña K, Sobraske PJ, Molina PA, Brookes PS, Galy V, Nehrke et al. Fndc-1 contributes to paternal mitochondria elimination in C. elegans. Dev Biol. 2019;454(1):15–20. 10.1016/j.ydbio.2019.06.016.
15. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014; 24(7):787–95. 10.1038/cr.2014.75.
16. Poole LP, Macleod KF. Mitophagy in tumorigenesis and metastasis. Cell Mol Life Sci. 2021;78(8):3817–51. 10.1007/s00018-021-03774-1.
17. Liu L, Feng D, Chen G, Chen M, Zheng Q, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–85. 10.1038/ncb2422.
18. Pan P, Chen J, Liu X, Fan J, Zhang D, Zhao W, et al. FUNDC1 regulates autophagy by inhibiting ROS-NLRP3 signaling to avoid apoptosis in the lung in a lipopolysaccharide-induced mouse model. Shock. 2021; 56(5):773–81. 10.1097/shk.0000000000001835..
19. Chang CC, Huang TY, Chen HY, Huang TC, Lin LC, Chang YJ, et al. Protective effect of melatonin against oxidative stress-induced apoptosis and enhanced autophagy in human retinal pigment epithelium cells. Oxid Med Cell Longev. 2018;2018:9015765. 10.1155/2018/9015765.
20. Chhabra R, Rao S, Kumar BM, Shetty AV, Hegde AM, Bhandary M. Characterization of stem cells from human exfoliated deciduous anterior teeth with varying levels of root resorption. J Clin Pediat Dentistry. 2021;45(2):104–11. 10.17796/1053-4625-45.2.6.
21. Fu Y, Jin R-R, Li Y-L, Luan H, Huang T, Zhao Y, et al. Isocorydine inhibits the proliferation of human endometrial carcinoma HEC-1B cells by downregulating the Ras/MEK/ERK signaling pathway. EJGO. 2021;42(3):548–53. 10.31083/j.ejgo.2021.03.5225.
22. Song C, Adili A, Kari A, Abuduhaer A. FSTL1 aggravates sepsis--induced acute kidney injury through regulating TLR4/MyD88/NF-κB pathway in newborn rats. Signa Vitae. 2021;17(3): 167–73. 10.22514/sv.2021.071.
23. Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J Pineal Res. 2017; 63(4):e12438. 10.1111/jpi.12438.
24. Morrison HA, Liu Y, Eden K, Nagai-Singer MA, Wade PA, Allen IC. NLRX1 deficiency alters the gut microbiome and is further exacerbated by adherence to a gluten-free diet. Front Immunol. 2022;13:882521. 10.3389/fimmu.2022.882521.
25. Pickering RJ, Booty LM. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology. 2021;162(3):268–80. 10.1111/imm.13291.
26. Yang Y, Yang X, Wu Y, Fu M. METTL3 promotes inflammation and cell apoptosis in a pediatric pneumonia model by regulating EZH2. Allergol Immunopathol (Madr). 2021;49(5): 49–56. 10.15586/aei.v49i5.445.
27. Martinez-Jaramillo C, Trujillo-Vargas CM. Dissecting the localization of lipopolysaccharide-responsive and beige-like anchor protein (LRBA) in the endomembrane system. Allergol Immunopathol (Madr). 2020;48(1):8–17. 10.1016/j.aller.2019.07.011.
28. Forman HJ, Zhang H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat Rev Drug Discov. 2021;20(9):689–709. 10.1038/s41573-021-00233-1.
29. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–76. 10.1056/NEJMra2022774.
30. Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration—Emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38(7):450–71. 10.1080/07853890600946724.
31. Khoo HE, Ng HS, Yap WS, Goh HJH, Yim HS. Nutrients for prevention of macular degeneration and eye-related diseases. Antioxidants (Basel). 2019;8(4):85. 10.3390/antiox8040085.
32. Hwang N, Kwon MY, Woo JM, Chung SW. Oxidative stress-induced pentraxin 3 expression human retinal pigment epithelial cells is involved in the pathogenesis of age-related macular degeneration. Int J Mol Sci. 2019;20(23):6028. 10.3390/ijms20236028. PMid: 31795454.
33. Lazzarini R, Nicolai M, Lucarini G, Pirani V, Mariotti C, Bracci M, et al. Oxidative stress in retinal pigment epithelium impairs stem cells: A vicious cycle in age-related macular degeneration. Mol Cell Biochem. 2022;477(1):67–77. 10.1007/s11010-021-04258-3..
34. Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy.2014;10(11):1989–2005.10.4161/auto.36184.
35. Hyttinen J, Blasiak J, Tavi P, Kaarniranta K. Therapeutic potential of PGC-1α in age-related macular degeneration (AMD)—The involvement of mitochondrial quality control, autophagy, and antioxidant response. Expert Opin Ther Targets. 2021;25(9):773–85. 10.1080/14728222.2021.1991913.
36. Ma D, Zhao Y, She J, Zhu Y, Zhao Y, Liu L, et al. NLRX1 alleviates lipopolysaccharide-induced apoptosis and inflammation in chondrocytes by suppressing the activation of NF-κB signaling. Int Immunopharmacol. 2019;71:7–13. 10.1016/j.intimp.2019.03.001.
37. Leber A, Hontecillas R, Zoccoli-Rodriguez V, Bienert C, Chauhan J, Bassaganya-Riera J. Activation of NLRX1 by NX-13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4(+) T cells. J Immunol. 2019;203(12): 3407–15. 10.4049/jimmunol.1900364.
38. Aikawa C, Nakajima S, Karimine M, Nozawa T, Minowa-Nozawa A, Toh H, et al. NLRX1 negatively regulates group A streptococcus invasion and autophagy induction by interacting with the Beclin 1-UVRAG complex. Front Cell Infect Microbiol. 2018;8:403. 10.3389/fcimb.2018.00403.
39. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–87. 10.1038/nm.3893.
40. Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, Knight DA, et al. Inflammasomes in the lung. Mol Immunol. 2017;86:44–55. 10.1016/j.molimm.2017.01.014.
Supplementary
Figure S1 Levels of SOD and GSH after ARPE-19 cells were treated with H2O2 or overexpressed with NLRX1. The concentrations of SOD and GSH were measured using commercial kits. The experiments were performed with three replicates, and the data were expressed as mean ± SD.
Figure S2 Concentrations of IL-1β, TNF-α, MCP-1, and IL-6 after ARPE-19 cells treated with H2O2 or overexpressed with NLRX1. The concentrations of IL-1β, TNF-α, MCP-1, and IL-6 were determined by commercial ELISA kits. The experiments were performed with three replicates, and the data were expressed as mean ± SD.
Figure S3 The relative protein expression of NLRP3 was examined by Western blot analysis through NLRP3 antibody (15101, CST).