
Download
ORIGINAL ARTICLE
Knockdown of EDA2R alleviates hyperoxia-induced lung epithelial cell injury by inhibiting NF-κB pathway
Nan Jiaa#, Yi Jiaa#, Fen Yanga*, Wenchao Dub
aDepartment of Neonatology, The Second Affiliated Hospital of Xi’an Medical College, Xi’an City, Shaanxi Province, China
bDepartment of Proctology, The Second Affiliated Hospital of Xi’an Medical College, Xi’an City, Shaanxi Province, China
Abstract
Background: Long-term hyperoxia impairs growth of the lungs and contributes to development of bronchopulmonary dysplasia. Ectodysplasin A (EDA) binds to ectodysplasin A2 receptor (EDA2R) and is essential for normal prenatal development. The functioning of EDA2R in bronchopulmonary dysplasia is investigated in this study.
Methods: Murine lung epithelial cells (MLE-12) were exposed to hyperoxia to induce cell injury. Cell viability and apoptosis were detected, respectively, by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay and flow cytometry. Inflammation and oxidative stress were evaluated by enzyme-linked immunosorbent serologic assay.
Results: Hyperoxia decreased cell viability and promoted cell apoptosis of MLE-12. EDA2R was elevated in hyperoxia-induced MLE-12. Silencing of EDA2R enhanced cell viability and reduced cell apoptosis of hyperoxia-induced MLE-12. Hyperoxia-induced up-regulation of tumor necrosis factor alpha (TNF-α), Interleukin (IL)-1β, and IL-18 as well as MLE-12 was suppressed by knockdown of EDA2R. Inhibition of EDA2R down-regulated the level of malondialdehyde (MDA), up-regulated superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH) in hyperoxia-induced MLE-12. Interference of EDA2R attenuated hyperoxia-induced increase in p-p65 in MLE-12.
Conclusion: Knockdown of EDA2R exerted anti-inflammatory and antioxidant effects against hyperoxia-induced injury in lung epithelial cells through inhibition of nuclear factor kappa B (NF-κB) pathway.
Key words: bronchopulmonary dysplasia, EDA2R, hyperoxia, inflammation, lung epithelial cells, NF-κB pathway, oxidative stress
*Corresponding author: Fen Yang, Department of Neonatology, The Second Affiliated Hospital of Xi’an Medical College, No. 167, Fangdong Street Textile City, Baqiao District, Xi’an City, Shanxi Province 710038, China. Email address: [email protected]
#These authors contributed equally to the work and should be considered first coauthors.
Received 16 May 2022; Accepted 6 June 2022; Available online 1 September 2022
Copyright: Jia N, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Hyperoxia is often administered to premature babies, and a long-term exposure to hyperoxia potentially induces oxygen toxicity, chronic lung injury, and bronchopulmonary dysplasia.1 Bronchopulmonary dysplasia induces arrest of vascular and alveolar development, and refers to common cause of respiratory disease with leading mortality and morbidity rates.2 Bronchopulmonary dysplasia is characterized by fibrosis, abnormal lung function, mesenchymal cell hyperplasia, pulmonary growth arrest, and enlarged alveoli.3 Several therapeutic strategies, such as Interleukin (IL)-1 receptor antagonist, surfactant therapy, and antenatal glucocorticoids, are currently used in the treatment of bronchopulmonary dysplasia.4 However, the limited clinical efficiency of these therapies raises prospects of novel therapies for bronchopulmonary dysplasia.
Long-term exposure to hyperoxia induces alveolar endothelial and epithelial cell death through necrosis or apoptosis.4 Moreover, hyperoxia could also stimulate production of reactive oxygen species (ROS), and promote expression of proinflammatory cytokines in alveolar epithelial cells, thus exacerbating lung injury and cell death in the development of bronchopulmonary dysplasia.4 Therefore, anti-oxidant and anti-inflammatory therapies might be regarded as promising treatment for bronchopulmonary dysplasia.5
Ectodysplasin-A2 receptor (EDA2R) is a tumor necrosis factor receptor (TNFR) that binds to ectodysplasin A (EDA) ligand and recruits intracellular adapter proteins to participate in normal prenatal development.6 EDA2R is associated with diabetic nephropathy, metabolic diseases, and tumor-igenesis through various signaling pathways.6 For example, EDA2R was involved in isoform EDA-A2-induced apoptosis of hair follicle cells7 and osteosarcoma cells8 through activation of cleaved caspase-3. EDA2R was reduced in breast cancer,9 and functioned as a tumor suppressor in colorectal cancer through p53-regulated anoikis pathway.10 Moreover, EDA2R suppressed colonic stemness and epithelial repair, thus contributing to intestinal inflammation.11 Silencing of EDA2R inhibited high glucose-induced accumulation of reactive oxygen species and cell apoptosis of podocytes.12 Therefore, EDA2R might be involved in hyperoxia-induced lung epithelial cell injury through inflammatory and oxidant properties. A previous study has demonstrated that EDA2R was up-regulated in the lungs of hyperoxia-induced neonatal mouse.13 The effects of EDA2R on cell apoptosis, inflammation, and oxidative stress of hyperoxia-induced lung epithelial cells were investigated in this study.
Materials and methods
Cell culture, treatment, and transfection
Murine lung epithelial cells (MLE-12) were acquired from ATCC (Manassas, VA, USA) and cultured in Dulbecco’s modified eagle medium (DMEM)/F-12 with 2% fetal bovine serum (FBS), 10-nM hydrocortisone, and 10-nM β-estradiol (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C. In order to induce oxygen toxicity, MLE-12 cells were kept in hyperoxic condition with 85% O2 for 24 h. Cells in the control group were exposed to normoxic condition with 21% O2. For cell transfection, MLE-12 cells were seeded in 96-well plates and transfected with plasmid cloning DNA (pcDNA) vector, pcDNA–EDA2R, small hairpin negative control (shNC), shEDA2R (GenePharma, Suzhou, China) via lipofectamine 2000 (Thermo Fisher Scientific) under hyperoxic condition.
Cell viability and apoptosis assays
MLE-12 cells were seeded in 96-well plates and subjected to various transfections for 24 h. Cells were treated with MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide); Beyotime, Beijing, China) for 4 h, and incubated with dimethyl sulfoxide (DMSO). Absorbance at 490 nm was measured using microplate autoreader (Thermo Fisher Scientific). In order to detect cell apoptosis, MLE-12 was trypsinized and re-suspended in binding buffer of annexin V-fluorescein isothiocyanate (V-FITC)/propidium iodide (PI) staining kit (BD Bioscience, San Diego, CA, USA). Cells were treated with annexin V-FITC and PI, and analyzed under FACSCanto II flow cytometer (BD Bioscience).
Enzyme linked immunosorbent serologic assay (ELISA)
Cell culture supernatants of MLE-12 cells were collected via centrifugation at 500× g for 5 min. Levels of IL-1β (ab100704), IL-18 (ab216165), and TNF-α (ab100747) were detected by commercial ELISA kits (Abcam, Cambridge, MA, USA). MLE-12 cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Beyotime), and centrifuged at 12,000× g to collect supernatants. Levels of SOD, MDA, CAT, and GSH were also determined by ELISA kits.
Western blot analysis
Protein samples of MLE-12 were segregated by using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred on nitrocellulose membranes. Membranes were blocked in 5% dry milk, and incubated with primary antibodies: anti-EDA2R and anti-β-actin (1:2000), anti-Bax and anti-Bcl-2 (1:3000), anti-p-IκBα (p-nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha) and anti-IκBα (1:4000), and anti-p-p65 and anti-p65 (1:5000). The membranes were incubated with secondary antibodies (1:5000), and subjected to electrochemiluminescence (ECL) reagent kit (Beyotime). All antibodies were purchased from Abcam.
Statistical analysis
All the data were expressed as mean ± standard error of mean (SEM) and analyzed by Student’s t-test or oneway analysis of variance (ANOVA) with SPSS 21 software. Bonferroni correction was used to control family-wise error rate; P < 0.05 was considered as statistically significant.
Results
EDA2R was elevated in hyperoxia-induced lung epithelial cell
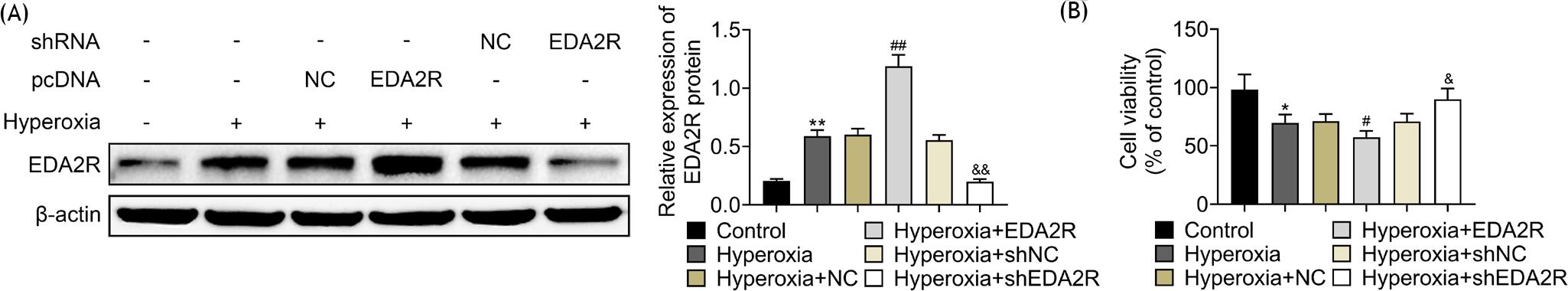
In order to induce oxygen toxicity, MLE-12 was kept in hyper-oxic condition. Protein expression of EDA2R was up-regulated in hyperoxia-induced MLE-12 (Figure 1A). Hyperoxic condition reduced cell viability of MLE-12 (Figure 1B). Hyperoxia-induced MLE-12 cells were then transfected with shEDA2R or pcDNA–EDA2R to decrease or increase EDA2R expression (Figure 1A). Over-expression of EDA2R decreased cell viability of hyperoxia-induced MLE-12 (Figure 1B), while silencing of EDA2R increased the cell viability of hyperoxia-induced MLE-12 (Figure 1B), suggesting the anti-proliferative effect of EDA2R against hyperoxia-induced lung epithelial cell.
Figure 1 EDA2R was elevated in hyperoxia-induced lung epithelial cell. (A) EDA2R was up-regulated in hyperoxia-induced MLE-12; transfection with shEDA2R or pcDNA-EDA2R decreased or increased EDA2R expression in hyperoxia-induced MLE-12. (B) Overexpression of EDA2R decreased cell viability of hyperoxia-induced MLE-12, while silencing of EDA2R increased cell viability of hyperoxia-induced MLE-12. *,**vs. control, P < 0.05, P < 0.01. #,##vs. hyperoxia + NC, P < 0.05, P < 0.01. &,&&vs. hyperoxia + shNC, P < 0.05, P < 0.01.
EDA2R promoted cell apoptosis of hyperoxia-induced lung epithelial cell
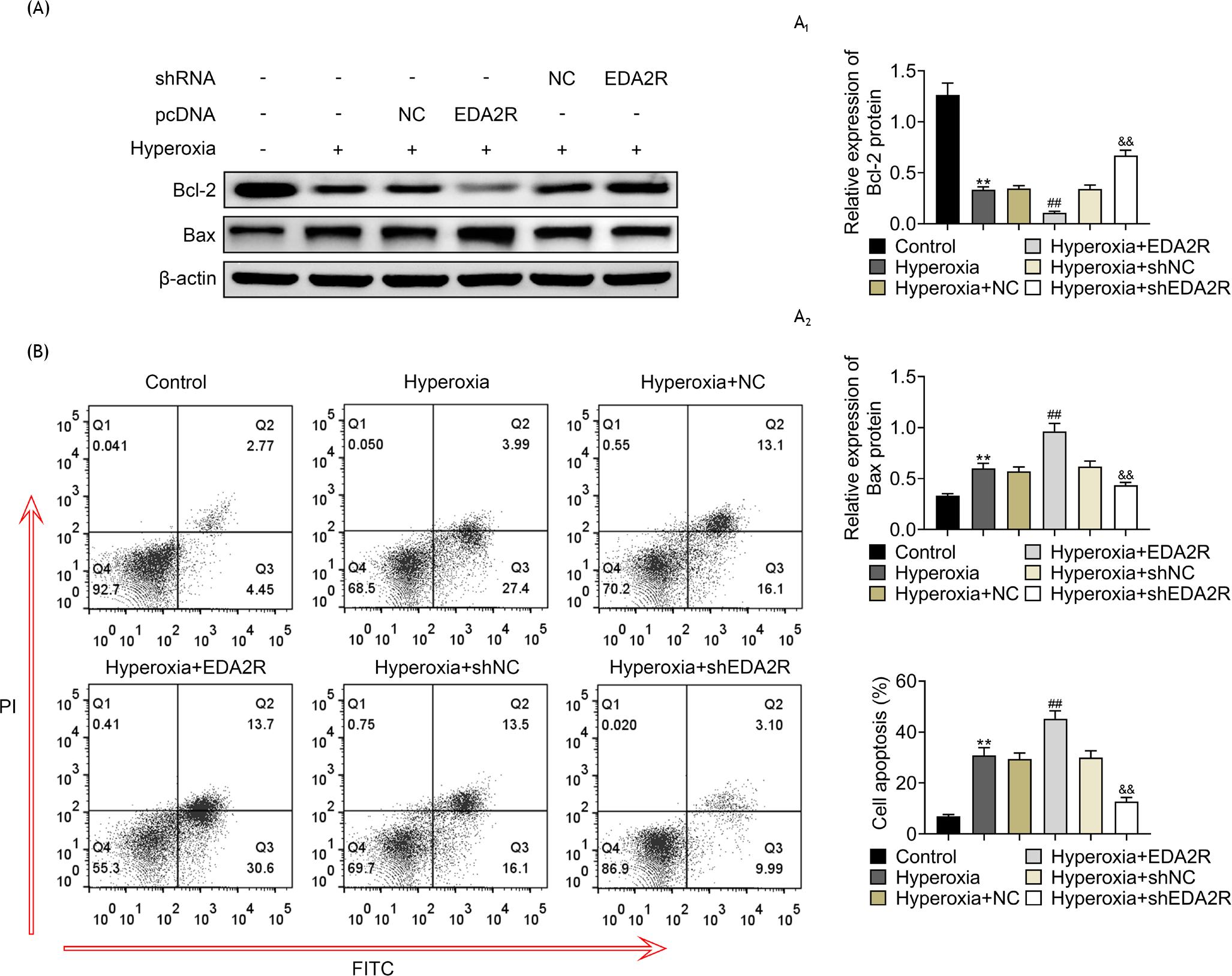
Protein expression of Bax was up-regulated, while Bcl-2 was down-regulated in MLE-12 by hyperoxic condition (Figures 2A, A1, and A2). Overexpression of EDA2R reduced Bcl-2 and enhanced Bax in hyperoxia-induced MLE-12 (Figures 2A, A1, and A2). Moreover, silencing of EDA2R reduced Bax and enhanced Bcl-2 in hyperoxia-induced MLE-12 (Figures 2A, A1, and A2). Overexpression of EDA2R promoted cell apoptosis of hyperoxia-induced MLE-12 (Figure 2B). Knockdown of EDA2R attenuated hyperoxia-induced increase in cell apoptosis in MLE-12 (Figure 2B), revealing pro-apoptotic effect of EDA2R against hyperoxia-induced lung epithelial cell.
Figure 2 EDA2R promoted cell apoptosis of hyperoxia-induced lung epithelial cell. (A) Overexpression of EDA2R reduced Bcl-2 and enhanced Bax in hyperoxia-induced MLE-12 (Figures 2A, A1, and A2); silencing of EDA2R reduced Bax and enhanced Bcl-2 in hyperoxia-induced MLE-12. (B) Overexpression of EDA2R promoted cell apoptosis of hyperoxia-induced MLE-12; knockdown of EDA2R inhibited cell apoptosis of hyperoxia-induced MLE-12. **vs. control, P < 0.01. ##vs. hyperoxia + NC, P < 0.01. &&vs. hyperoxia + shNC, P < 0.01.
EDA2R promoted inflammation of hyperoxia-induced lung epithelial cell
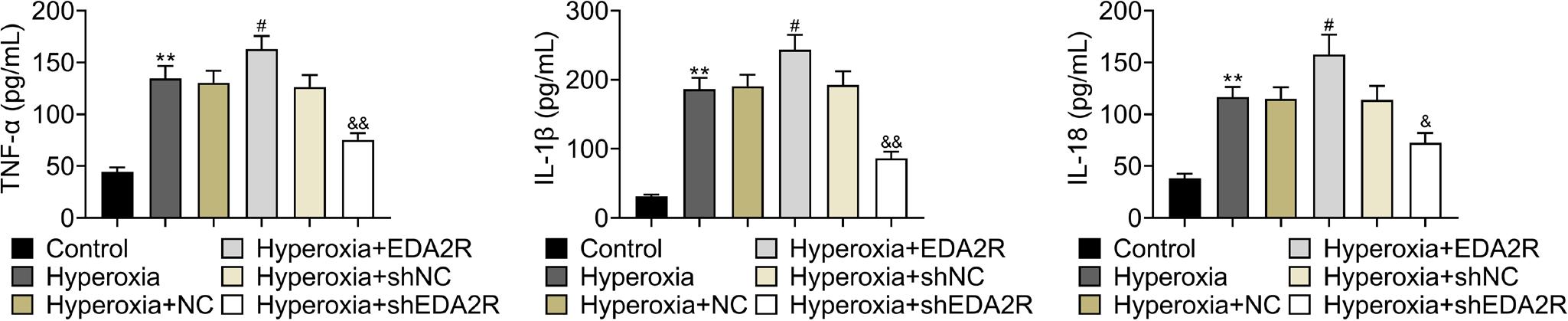
Hyperoxia induced up-regulation of IL-1β, IL-18, and TNF-α in MLE-12 (Figure 3). Overexpression of EDA2R aggravated hyperoxia-induced inflammation in MLE-12 by further up-regulation of IL-1β, IL-18, and TNF-α (Figure 3). However, loss of EDA2R exerted anti-inflammatory effect on hyper-oxia-induced MLE-12 through down-regulation of IL-1β, IL-18, and TNF-α (Figure 3).
Figure 3 EDA2R promoted inflammation of hyperoxia-induced lung epithelial cell. Overexpression of EDA2R up-regulated levels of IL-1β, IL-18, and TNF-α in hyperoxia-induced MLE-12; loss of EDA2R down-regulated IL-1β, IL-18, and TNF-α in hyperoxia-induced MLE-12. **vs. control, P < 0.01. #vs. hyperoxia + NC, P < 0.05. &,&&vs. hyperoxia + shNC, P < 0.05, P < 0.01.
EDA2R promoted oxidative stress of hyperoxia-induced lung epithelial cell
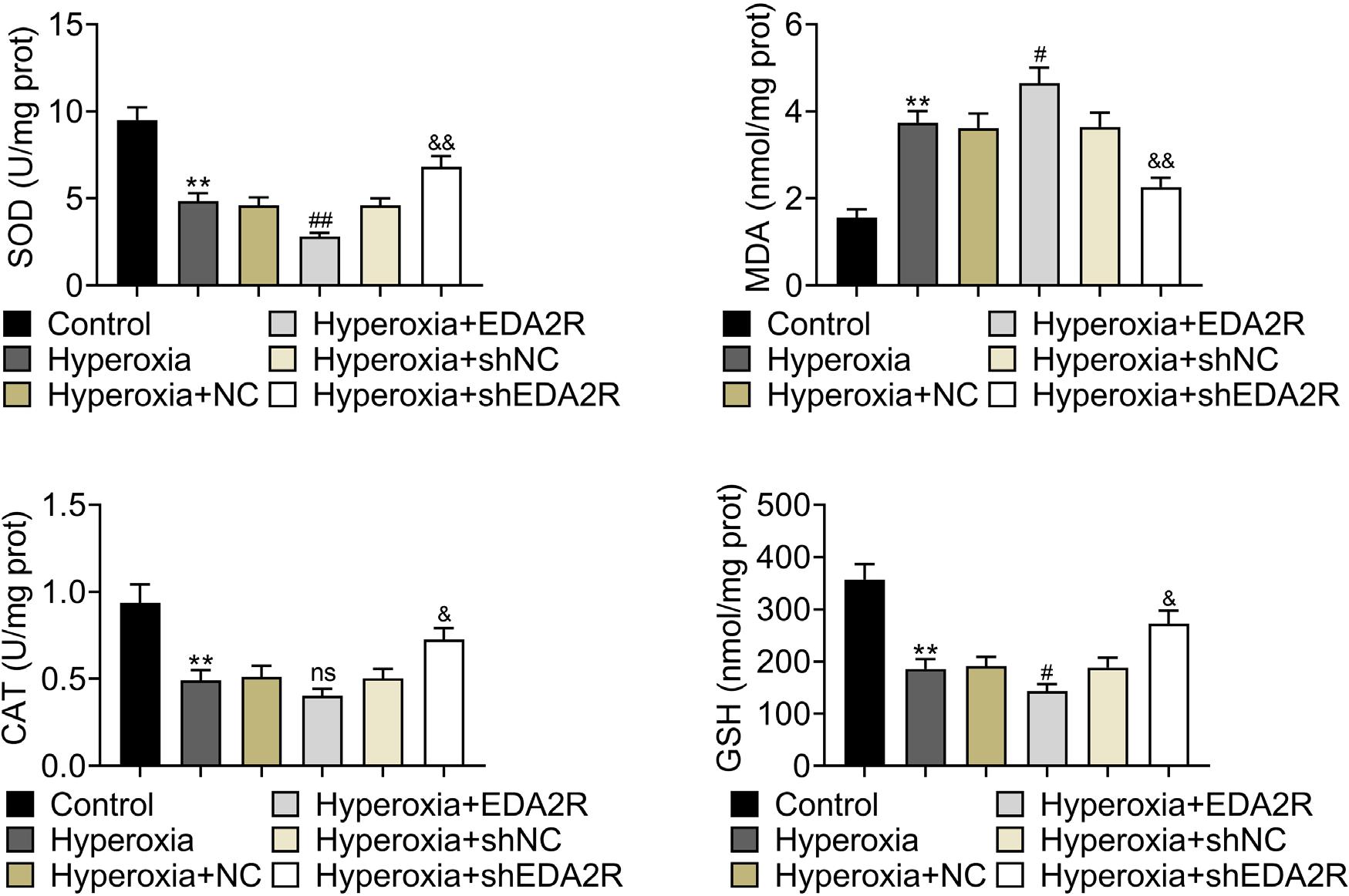
Level of MDA in MLE-12 was enhanced, while that of SOD, CAT, and GSH were reduced by hyperoxic condition (Figure 4). Interference of EDA2R reduced MDA but enhanced SOD, CAT, and GSH in hyperoxia-induced MLE-12 (Figure 4). Moreover, overexpression of EDA2R promoted oxidative stress of hyperoxia-induced MLE-12 by increasing MDA but decreasing SOD, CAT, and GSH (Figure 4).
Figure 4 EDA2R promoted oxidative stress of hyperoxia-induced lung epithelial cell. Overexpression of EDA2R increased level of MDA, and decreased SOD, CAT, and GSH in hyperoxia-induced MLE-12; interference of EDA2R reduced MDA, and enhanced SOD, CAT, and GSH in hyperoxia-induced MLE-12. **vs. control, P < 0.01. #vs. hyperoxia + NC, P < 0.05. &, &&vs. hyperoxia + shNC, P < 0.05, P < 0.01.
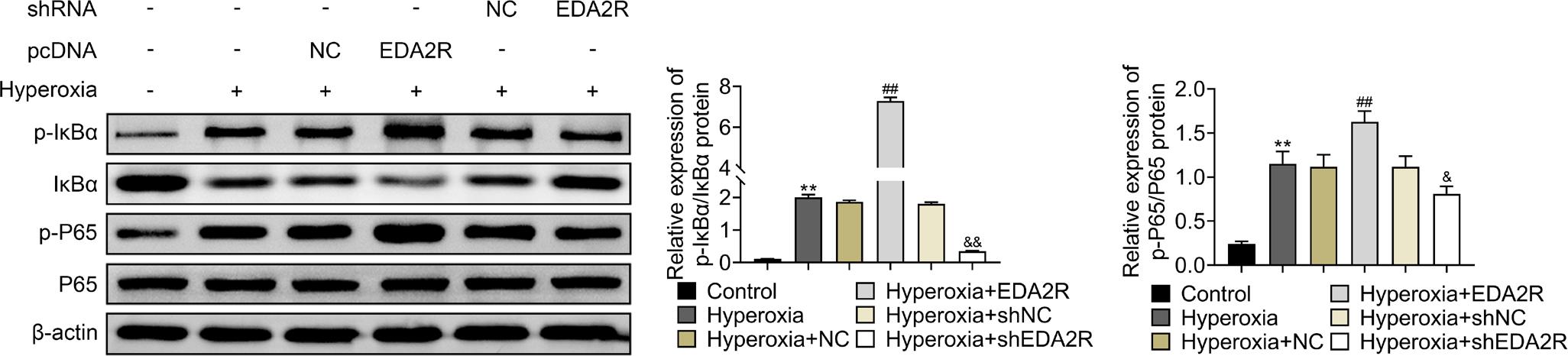
EDA2R promoted activation of nuclear factor kappa B (NF-κB) signaling in hyperoxia-induced lung epithelial cell
Overexpression of EDA2R aggravated hyperoxia-induced decrease of IκBα expression but increased p-IκBα and p-p65 expression in MLE-12 (Figure 5). However, silencing of EDA2R increased protein expression of IκBα but decreased p-IκBα and p-p65 expression in hyperoxia-induced MLE-12 (Figure 5).
Figure 5 EDA2R promoted activation of NF-κB signaling in hyperoxia-induced lung epithelial cell. Overexpression of EDA2R decreased protein expression of IκBα, increased p-IκBα and p-p65 in hyperoxia-induced MLE-12, silencing of EDA2R increased protein expression of IκBα, decreased p-IκBα and p-p65 in hyperoxia-induced MLE-12. **vs. control, P < 0.01. ##vs. hyperoxia + NC, P < 0.01. &,&&vs. hyperoxia + shNC, P < 0.05, P < 0.01.
Discussion
EDA–EDAR axis activates multiple signaling pathways, such as fibroblast growth factor pathway, c-Jun N-terminal kinase pathway, bone morphogenetic protein pathway, and Wnt/β-catenin signaling, to regulate hair development and skeletal muscle homeostasis.6 EDA2R was reported to be the the most highly induced gene in hyperoxia-induced neonatal mouse lungs.13 This study, for the first time, determined that EDA2R was involved in hyperoxia-induced lung epithelial cell injury by promoting inflammation and oxidative stress. Silencing of EDA2R reduced hyperoxia-induced inflammation and oxidative stress in lung epithelial cell, thus ameliorating progression of bronchopulmonary dysplasia.
A previous study has shown demonstrated that hyper-oxia promoted airway resistance and inhibited dynamic lung compliance, contributing to impairment of lung function.14 Therefore, hyperoxia-induced oxygen toxicity in MLE-12 was widely used to establish in vitro cell model of bronchopulmonary dysplasia.15 In this study, hyperoxic condition decreased cell viability and increased cell apoptosis in MLE-12. EDA2R was up-regulated in hyperoxia-induced MLE-12. Alveolar death by apoptosis or necrosis is a common feature of hyperoxia-induced toxicity in the lungs.16 Inhibition of hyperoxia-induced cell apoptosis was considered to be a promising strategy for the treatment of bronchopulmonary dysplasia.16 Inactivation of EDA2R conferred resistance of colorectal cancer cell to p53-induced apoptosis.10 This study also established that loss of EDA2R decreased protein expression of Bax and increased Bcl-2 to inhibit cell apoptosis of hyperoxia-induced MLE-12, suggesting the antiapoptotic effects of EDA2R silencing on bronchopulmonary dysplasia.
Inflammatory responses and oxidative damage are the primary manifestations of hyperoxia-induced lung injury.17 Hyperoxic condition stimulated accumulation of reactive oxygen species to promote oxidative stress, and the oxidative stress promoted secretion of proinflammatory cytokines, thus leading to inflammatory and oxidative damage in alveolar epithelial cells during development of broncho-pulmonary dysplasia.18 Suppression of oxidative stress and inflammatory response ameliorated hyperoxia-induced lung injury.18 EDA2R contributed to generation of reactive oxygen species in podocytes,12 and aggravated intestinal inflammation.11 Results of this study demonstrated that loss of EDA2R reduced levels of IL-1β, IL-18, and TNF-α in hyperoxia-induced MLE-12. Moreover, knockdown of EDA2R reduced MDA, and enhanced SOD, CAT, and GSH in hyper-oxia-induced MLE-12, indicating anti-inflammatory and anti-oxidant effects of EDA2R silencing on bronchopulmonary dysplasia.
NF-κB, essential for production of proinflammatory factors,19 is important for inflammatory response in hyperoxia-induced alveolar epithelial cells.20 Inactivation of NF-κB signaling protected the lungs against hyperoxia-induced injury.21 EDA-A2–EDA2R complex recruited tumor necrosis factor receptor-associated factor 6 (TRAF6) and promoted activation of IkappaB (IκB) kinase complex through phosphorylation of inhibitor of nuclear factor kappa B alpha (IκBα), thus facilitating nuclear translocation of NF-κB.22 This study revealed that knockdown of EDA2R decreased phosphorylation of IκBα and p65 in hyperoxia-induced MLE-12, suggesting that loss of EDA2R might suppress activation of NF-κB signaling to attenuate bronchopulmonary dysplasia.
Conclusions
EDA2R was elevated in hyperoxia-induced MLE-12, and knockdown of EDA2R reduced cell apoptosis of hyperoxia-induced MLE-12. Moreover, silencing of EDA2R exerted anti-inflammatory and antioxidant effects on hyperoxia-induced MLE-12 through inactivation of NF-κB signaling. However, as limitations to this study, we explored only the role of EDA2R in one cell line. The effect of EDA2R on in vitro model of hyperoxia-induced primary or human cells must be investigated in the future studies. In addition, the effect of EDA2R on animal model of hyperoxia-induced bronchopulmonary dysplasia should also be explored in the future investigation.
Competing interests
The authors stated that there were no conflicts of interest to disclose.
Ethics approval
This article does not contain any experiments involving human or animal participation by any of the authors.
Data availability
The authors declare that all data supporting the findings of this study are available in the paper, and any raw data can be obtained from the corresponding author upon request.
Author Contribution
Nan Jia and Yi Jia designed and conducted the study; they also supervised data collection, and analyzed and interpreted the collected data. Fen Yang and Wenchao Du prepared and reviewed the draft of manuscript for publication. All the authors read and approved the final manuscript.
REFERENCES
1. Liang Z, Zhang X, Liu Y, Wu Q, You C. SEMA3A protects against hyperoxia-induced lung injury in a bronchopulmonary dysplasia model of newborn rat by inhibiting ERK pathway. Allergologia et Immunopathol. 2021;49(6):8–15. 10.15586/aei.v49i6.478
2. Thébaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, et al. Bronchopulmonary dysplasia. Nat Rev Dis Primers. 2019;5(1):78–. 10.1038/s41572-019-0127-7
3. Kalikkot Thekkeveedu R, Guaman MC, Shivanna B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir Med. 2017;132:170–7. 10.1016/j.rmed.2017.10.014
4. Gien J, Kinsella JP. Pathogenesis and treatment of broncho-pulmonary dysplasia. Curr Opin Pediatr. 2011;23(3):305–13. 10.1097/MOP.0b013e328346577f
5. Nitkin CR, Bonfield TL. Balancing anti-inflammatory and anti-oxidant responses in murine bone marrow-derived macrophages. PLoS One. 2017;12(9):e0184469-e. 10.1371/journal.pone.0184469
6. Cai Z, Deng X, Jia J, Wang D, Yuan G. Ectodysplasin A/ectodysplasin A receptor system and their roles in multiple diseases. Front Physiol. 2021;12:788411–. 10.3389/fphys.2021.788411
7. Kwack M, Kim J, Kim M. Ectodysplasin-A2 induces apoptosis in cultured human hair follicle cells and promotes regression of hair follicles in mice. Biochem Biophys Res Commun. 2019;520(2):428–433. 10.1016/j.bbrc.2019.10.0318.
8. Chang B, Punj V, Shindo M, Chaudhary PM. Adenoviral-mediated gene transfer of ectodysplasin-A2 results in induction of apoptosis and cell-cycle arrest in osteosarcoma cell lines. Cancer Gene Ther. 2007;14(11):927–33. 10.1038/sj.cgt.7701078
9. Punj V, Matta H, Chaudhary PM. X-linked ectodermal dysplasia receptor is downregulated in breast cancer via promoter methylation. Clin Cancer Res. 2010;16(4):1140–8. 10.1158/1078-0432.CCR-09-2463
10. Tanikawa C, Furukawa Y, Yoshida N, Arakawa H, Nakamura Y, Matsuda K. XEDAR as a putative colorectal tumor suppressor that mediates p53-regulated anoikis pathway. Oncogene. 2009;28(34):3081–92. 10.1038/onc.2009.154
11. Song L, Chang R, Sun X, Lu L, Gao H, Lu H, et al. Macrophage-derived EDA-A2 inhibits intestinal stem cells by targeting miR-494/EDA2R/β-catenin signaling in mice. Commun Biol. 2021;4(1):213. 10.1038/s42003-021-01730-0
12. Lan X, Kumar V, Jha A, Aslam R, Wang H, Chen K, et al. EDA2R mediates podocyte injury in high glucose milieu. Biochimie. 2020;174:74–83. 10.1016/j.biochi.2020.04.003
13. McGrath-Morrow SA, Lauer T, Collaco JM, Lopez A, Malhotra D, Alekseyev YO, et al. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine. 2014;65(1):4–9. 10.1016/j.cyto.2013.09.021
14. Jiao B, Tang Y, Liu S, Guo C. Tetrandrine attenuates hyper-oxia-induced lung injury in newborn rats via NF-κB p65 and ERK1/2 pathway inhibition. Ann Transl Med. 2020;8(16):1018. 10.21037/atm-20-5573
15. Vohlen C, Mohr J, Fomenko A, Kuiper-Makris C, Grzembke T, Aydogmus R, et al. Dynamic regulation of GH–IGF1 signaling in injury and recovery in hyperoxia-induced neonatal lung injury. Cells. 2021;10(11):2947. 10.3390/cells10112947
16. Zhang Y, Du H, Yu X, Zhu J. Fucoidan attenuates hyperoxia-induced lung injury in newborn rats by mediating lung fibro-blasts differentiate into myofibroblasts. Ann Transl Med. 2020;8(22):1501–. 10.21037/atm-20-6601
17. Ratner V, Slinko S, Utkina-Sosunova I, Starkov A, Polin RA, Ten VS. Hypoxic stress exacerbates hyperoxia-induced lung injury in a neonatal mouse model of bronchopulmonary dysplasia. Neonatology. 2009;95(4):299–305. 10.1159/000178798
18. Wang X, Huo R, Liang Z, Xu C, Chen T, Lin J, et al. Simvastatin inhibits NLRP3 inflammasome activation and ameliorates lung injury in hyperoxia-induced bronchopulmonary dysplasia via the KLF2-mediated mechanism. Oxid Med Cell Longev. 2022;2022:8336070–. 10.1155/2022/8336070
19. Fuhai Chen WW, Xianguo Cai, Hongsheng Yu, Chunsheng Qu, Xianjun Zhang, Leili Lv, et al. Methyl jasmonate reduces the inflammation and apoptosis of HK-2 cells induced by LPS by regulating the NF-κB pathway. Signa Vitae. 2021;17(3):218–24.
20. Chen S, Wu Q, Zhong D, Li C, Du L. Caffeine prevents hyper-oxia-induced lung injury in neonatal mice through NLRP3 inflammasome and NF-κB pathway. Respir Res. 2020;21(1):140. 10.1186/s12931-020-01403-2
21. Liu D, Wang Y, Li L, Zhao H, Li L, Liu Y, et al. Celecoxib protects hyperoxia-induced lung injury via NF-κB and AQP1. Front Pediatr. 2019;7:228. 10.3389/fped.2019.00228
22. Yan M, Wang L-C, Hymowitz Sarah G, Schilbach S, Lee J, Goddard A, et al. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science. 2000;290(5491):523–7. 10.1126/science.290.5491.523