
Download
ORIGINAL ARTICLE
Sevoflurane ameliorates LPS-induced inflammatory injury of HK-2 cells through Sirtuin1/NF-κB pathway
Wang Peipeia, Wang Pinga, Yin Miaomiaoa, Wang Shuob, c*
aDepartment of Anesthesiology, The First People’s Hospital of Lianyungang/The Affiliated Lianyungang Hospital of Xuzhou Medical University, Lianyungang, Jiangsu Province, China
bDepartment of Anesthesiology, Wujin Hospital Affiliated with Jiangsu University, Changzhou, Jiangsu Province, China
cDepartment of Anesthesiology, The Wujin Clinical College of Xuzhou Medical University, Changzhou, Jiangsu Province, China
Abstract
The anesthetic sevoflurane (SEV) has been shown to protect against organ’s injury during sepsis. The present study intended to uncover the protective effects of SEV on sepsis-induced acute kidney injury (SI-AKI) and its possible mechanism. Human renal tubular epithelial cell HK-2 was treated with 10 μg/mL lipopolysaccharide (LPS) to construct SI-AKI cell model. LPS-induced HK-2 cells were pretreated with SEV in the absence or presence of EX527, an inhibitor of Sirtuin 1 (SIRT1), after which were the detection of cell viability, lactate dehydrogenase (LDH) release, apoptosis, inflammation, and oxidative stress. Our results demonstrated that LPS caused decreased cell viability, increased LDH release, improved cell apoptosis along with decreased expression of Bcl2 and enhanced expressions of Bax, cleaved PARP and cleaved caspase, enhanced production, and protein expressions of TNF-α, IL-6, and IL-1β, increased generation of reactive oxygen species (ROS) and malondialdehyde (MDA), but contributed to declined activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px). LPS inhibited SIRT1 and IκBα expressions but up-regulated p-NF-κB p65 and acetyl-p53 expressions as well. However, SEV pretreatment abolished all above-mentioned effects of LPS on HK-2 cells, while EX527 significantly reversed the effects of SEV. In conclusion, SEV effectively protected HK-2 cells against LPS-induced apoptosis, inflammation, and oxidative stress, and these effects may depend on the increase of SIRT1 expression, thereby inactivating NF-κB signaling.
Key words: Acute kidney injury, nuclear factor kappa-B, sepsis, sevoflurane, Sirtuin 1
*Corresponding author: Shuo Wang, Wujin Hospital Affiliated with Jiangsu University, No. 2 Yongning North Road, Changzhou City, Jiangsu Province 213000, China. Email address: [email protected]
Received 8 March 2022; Accepted 27 April 2022; Available online 1 July 2022
Copyright: Wang P, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Sepsis-induced acute kidney injury (SI-AKI), a common complication in critically ill patients, is associated with high morbidity and mortality.1 Acute kidney injury (AKI) is now considered as a systemic disease, which is characterized by rapid decline in renal function as well as high morbidity and mortality.2 Owing to the lack of effective and reliable therapeutic interventions to prolong survival, relieve injury, or improve recovery, AKI remains a major medical problem.3 Although acute renal tubular necrosis, oxidative stress, inflammation, and apoptosis are related to AKI, the molecular mechanism is still poorly understood.4 Understanding the underlying mechanisms of AKI is of great clinical importance.
Sevoflurane (SEV) is a volatile anesthetic that is widely applied for the induction and maintenance of general anesthesia. In addition to its anesthetic properties, SEV has been reported to inhibit inflammation and apoptosis during sepsis to protect against organ’s injury including brain,5,6 lung,7 heart,8 and liver.9 For example, SEV was found to exert brain-protective effects on sepsis via regulating apoptosis-related signaling pathway in a rat model with sepsis.5 SEV showed a cytoprotective effect in human umbilical vein endothelial cells with LPS stimulation through decreasing cell death and reducing the releases of the inflammatory mediators TNF-α and IL-6.10 Comparing the effects of SEV and propofol on AKI in children with living donor liver transplantation, it was found that SEV has a link to the modest decrease in AKI incidence when compared with propofol,11 suggesting the potential value of SEV in relieving AKI. However, the effects of SEV on SI-AKI remain elusive.
Sirtuin 1 (SIRT1), a nuclear protein and a key member of the class III histone deacetylase (HDAC) family and the function of SIRT1 depends on nicotinamide-adenine dinucleotide (NAD). Due to the important role of NAD + in energy metabolism and aging, SIRT1 has been shown to play an important role in many biological processes, including glucose and lipid metabolism, autophagy, inflammation, oxidative stress, apoptosis, and other processes.12–15 It has been implicated to attenuate sepsis-associated organ injury including SI-AKI.16,17 SIRT1 is widely expressed in various tissues, while SIRT1 mainly exists in mesangial cells and epithelial cells of renal tubules in the kidney.18 In vitro experiments show that SIRT1 can be used in the protection of kidney injury induced by oxidant, cisplatin, and ischemia-reperfusion through antioxidant stress, mitochondrial regeneration, and inhibition of apoptosis.19–21 Notably, recent studies have reported that SEV exerted its effect through increasing SIRT1 expression.6,22 SEV exhibited therapeutic potential for the treatment of septic encephalopathy and related cognitive malfunction via up-regulating SIRT1 in a sepsis model.6 The SEV-mediated protective effects on myocardial ischemia-reperfusion injury were also associated with SIRT1 expression.22 In this study, we intended to clarify the protective effects of SEV on SI-AKI in vitro using LPS-induced HK-2 cell model, and to uncover whether SIRT1 played a key role in this effect.
Materials and Methods
Cell culture and treatment
Chinese Academy of Science Cell Bank (Shanghai, China) provided the human renal tubular epithelial cell line HK-2 which was cultured in Keratinocyte Serum Free Medium (K-SFM; Invitrogen, 17005-042) supplemented with 10% FBS and Gentamicin/Amphotericin Solution 500× (Gibco). The cells were maintained in an incubator at 37°C with 5% CO2. For the establishment of SI-AKI cell model, HK-2 cells were stimulated with 10 μg/mL LPS (Sigma) for 24 h.23 For SEV treatment, cells were pre-treated with 2.4% SEV (Sigma) for 1 h.9,24 To inhibit SIRT1, LPS plus SEV-treated HK-2 cell line was co-treated with 10 μM EX527 (MedChem Express), an inhibitor of SIRT1, for 24 h.25
Cell viability measurement
Cell viability was determined by a cell counting kit-8 (CCK-8) assay (Beyotime). Briefly, the HK-2 cells (3×103/well) were inoculated into a 96-well plate. After the cells were cultured for 24 h in a normal medium, the medium was replaced with a new medium according to different group intervention conditions. After being cultured for another 24 h, the cells in each well were exposed to 10 μL of CCK-8 solution at 37°C in the dark for 2 h. The absorbance value of each well at 450 nm wavelength was measured using a microplate reader (Thermo Scientific Multiskan™ FC).
Lactate dehydrogenase (LDH) release detection
The relative level of LDH release was determined by an LDH assay kit (#ab65393; Abcam). Briefly, the HK-2 cell line was seeded into 96-well plates (4×104 cells/well) and then cultured in a complete medium with or without LPS, SEV, and EX527. After 24 h of incubation, the cell culture medium supernatants were collected by centrifuging the culture medium at 600 g for 10 min. After transferring the supernatants into another 96-well plate, each well was added with 100 μL LDH reaction mix and incubated at room temperature for 0.5 h. The optical density at 490 nm was determined using a microplate reader (Thermo Scientific Multiskan™ FC).
TUNEL staining
Cell apoptosis was evaluated by a TUNEL Assay Kit (Beyotime, China) according to manufacturer’s protocol. In brief, after being cultured in different mediums for 24 h, the HK-2 cell line was fixed with 1% paraformaldehyde, followed by the incubation with DNA labeling solution at 37°C for 1 h. After the addition of PI/RNase A solution for 0.5 h, TUNEL-positive (green) cells were captured using a fluorescent microscope (Zeiss L LSM800) (magnification ×200). Apoptotic cells (%) = the number of apoptotic cells/the number of total cells. The quantification of apoptotic cells was determined by Image J 1.51 software (National Institutes of Health) in five fields.
Western blotting assay
Total proteins from HK-2 cells were extracted on ice using a RIPA lysis buffer (Transgene, China) and then quantified by a BCA kit (Transgene). Equal amount of proteins (30 μg) were subjected to 10–12% SDS-PAGE for separation and then transferred onto PVDF membranes (Millipore, USA). After being incubated with 5% non-fat milk for 1.5 h and probed with primary antibodies overnight at 4°C, membranes were probed with a goat anti-rabbit IgG secondary antibody (#ab6721, 1:10000, Abcam). The densities of bands were detected with ECL assay (Thermo Fisher) and analyzed with Image J software. The rabbit primary antibodies (all from Abcam) included Bcl-2 (#ab32124, 1:1000), Bax (#ab32503, 1:5000), cleaved PARP (#ab32064, 1:5000), cleaved caspase 3 (#ab32042, 1:500), TNF-α (#ab183218, 1:1000), IL-6 (#ab233706, 1:1000), IL-1β (#ab254360, 1:1000), SIRT1 (#ab189494, 1:1000), IκBα (#ab32518, 1:5000), phosphorylated (p)-NF-κB p65 (#ab76302, 1:1000), NF-κB p65 (#ab32536, 1:5000), acetyl-p53 (#ab75754, 1:5000), p53 (#ab32389, 1:10000), and GAPDH (#ab9485, 1:10000).
ELISA
The secretion of inflammatory cytokines including TNF-α, IL-6, and IL-1β was measured in a cell supernatant using respective ELISA kits against TNF-α (#ab181421, Abcam), IL-6 (#ab178013, Abcam), and IL-1β (#ab100562, Abcam) strictly in accordance with manufacturer’s instructions.
Reactive oxygen species (ROS) production
The production of ROS was observed using a diacetate 2',7'-dichlorofluorescein diacetate (DCFDA) Cellular ROS Kit (ab113851, Abcam) at 37°C in the dark, strictly in accordance with manufacturer’s guide. Briefly, HK-2 cells were seeded into 96-well plates (4×104 cells/well) and cultured in normal medium overnight for adherence. After exposure to different drugs, cells were stained with DCFDA for 45 min, and then the ROS level (DCF fluorescent) was detected by fluorescence microscope (Zeiss L LSM800, Germany) (magnification ×200) with excitation/emission at 485 nm/535 nm.
Detection of malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px)
The concentration of MDA and activity of SOD and GSH-Px in HK-2 cells were tested using a Lipid Peroxidation (MDA) Assay Kit (Abcam, ab118970), SOD Assay Kit (Abcam, ab65354), and GSH-Px Assay Kit (Abcam, ab102530), respectively, according to manufacturer’s protocol.
Statistical analysis
All data were analyzed using SPSS 19.0 (IBM Corp.) and GraphPad Prism 8.0 (GraphPad Software, Inc.). All data were detected by Shapiro–Wilk (S–W) to evaluate whether they fit the normal distribution. If the data conformed to the normal distribution, comparisons among different groups were calculated using one-way ANOVA followed by Tukey’s post-hoc test. If the data did not conform to the normal distribution, the non-parametric test statistical analysis method was adopted. P < 0.05 was thought to indicate significance.
Results
SEV protects HK-2 cells against LPS-induced cytotoxicity and apoptosis, whereas SIRT1 inhibition blocks this effect
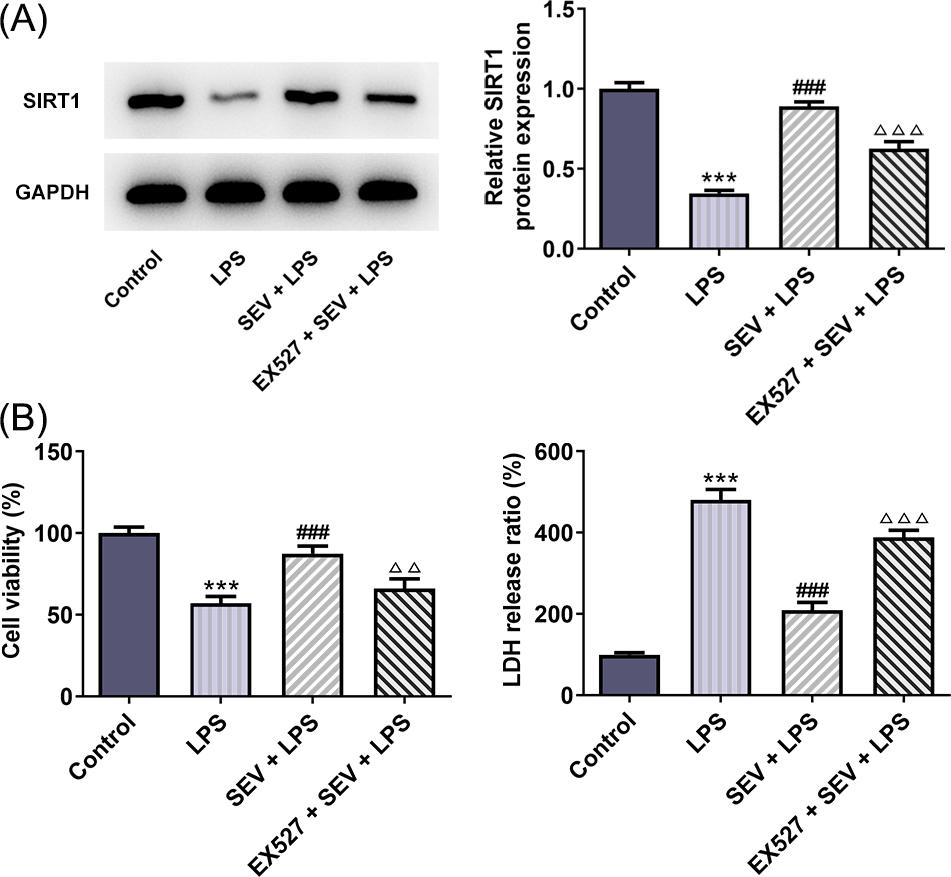
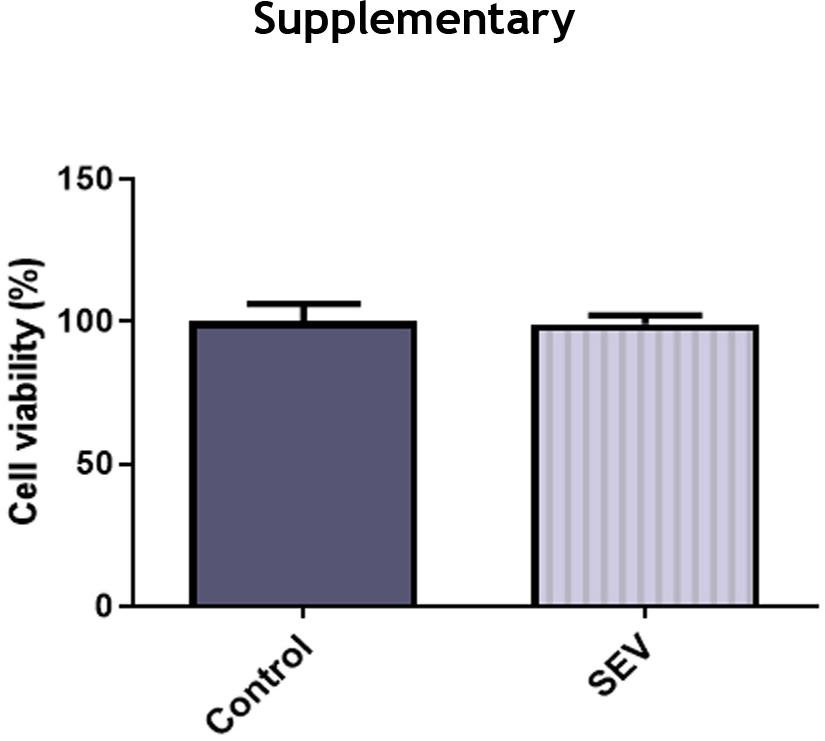
To investigate the effect of SEV on HK-2 cells, the viability of HK-2 cells was detected by CCK-8 assay and the result indicated that SEV did not affect HK-2 cells (Figure S1). To investigate the effect of SEV on LPS-induced HK-2 injury, HK-2 cells that were stimulated with LPS were pre-treated with SEV or not. To uncover whether SEV exerted its effects on LPS-induced HK-2 injury through up-regulating SIRT1 expression, an inhibitor of SIRT1, EX527 was utilized to challenge HK-2 cells that were exposed to SEV + LPS. First of all, cell viability and cytotoxicity were measured by CCK-8 and LDH release assays. As shown in Figure 1(A), LPS treatment considerably reduced cell viability (56.9%) of control HK-2 cells. However, SEV pre-treatment markedly increased the cell viability (87.4%) of LPS-treated HK-2 cells. Moreover, compared with SEV + LPS, the additional presence of EX527 decreased HK-2 cells viability (66.0%). Figure 1(B) revealed that LPS caused a sharp increase in LDH release in HK-2 cells, suggesting the strong cytotoxicity caused by LPS. On the contrary, SEV inhibited LDH release of LPS-induced HK-2 cells, which was reversed by EX527 co-treatment. These data showed that the cell viability impairment and cytotoxicity caused by LPS were relieved by SEV and this may depend on up-regulating SIRT1 expression.
Figure 1 SEV prevents LPS-induced cytotoxicity in HK-2 cells through activating SIRT1 expression. (A) CCK-8 assay was used to detect the cell viability of HK-2 cells in different groups. (B) LDH release kit was used to detect LDH release, data were shown as % of control group. N=3. ***P<0.001 vs. control; ###P<0.001 vs. LPS; ∆∆P<0.01 and ∆∆∆P<0.001 vs. SEV+LPS.
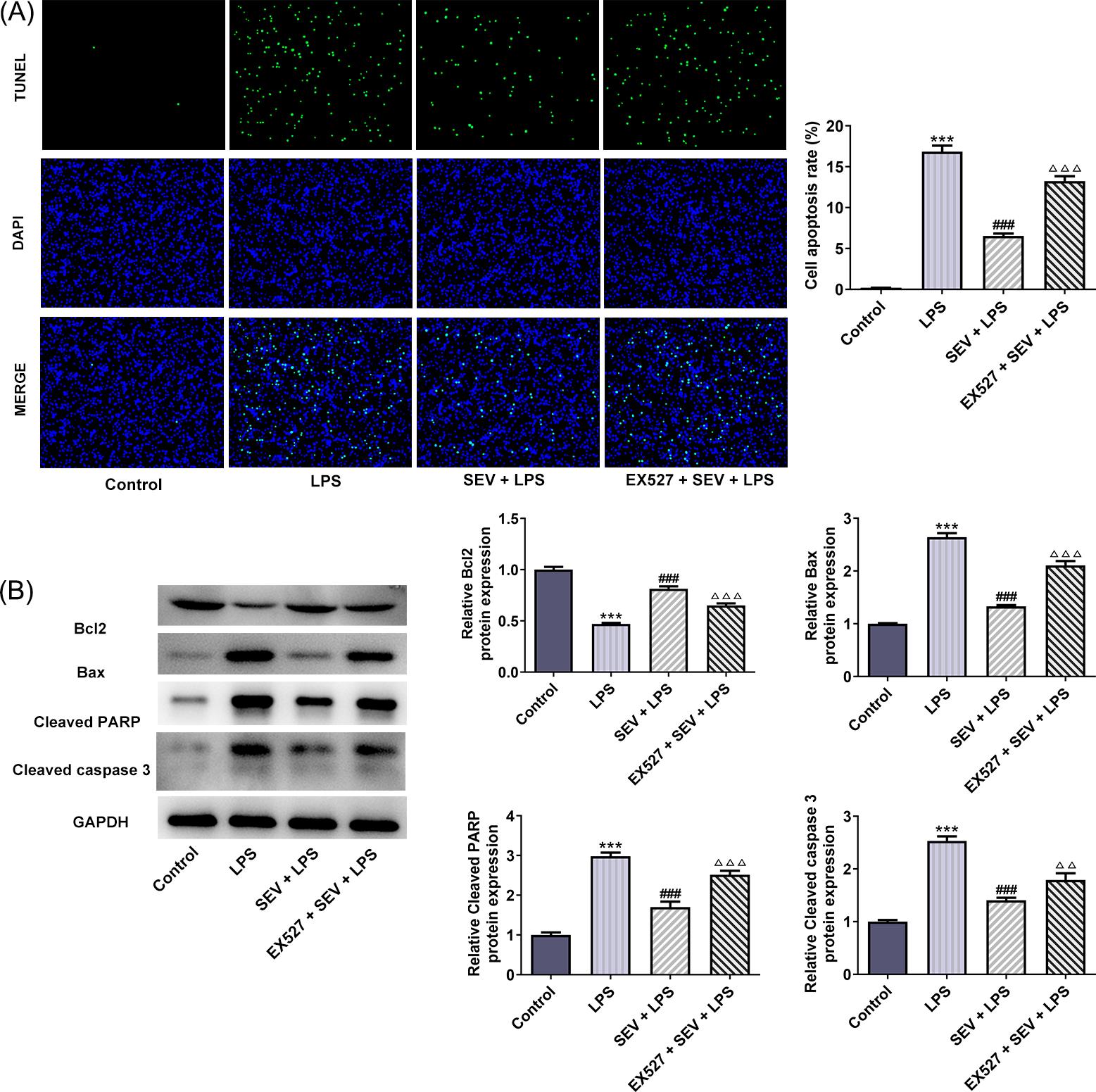
Next, cell apoptosis was evaluated. Figure 2(A) illustrated the TUNEL staining of HK-2 cells in different groups. It can be observed that LPS resulted in a remarkable increase in TUNEL-positive cells (green), suggesting the occurrence of apoptosis induced by LPS. However, apoptosis was significantly reduced in cells pretreated with SEV compared with LPS-induced HK-2 cells, while cells treated with EX527 + SEV + LPS exhibited increased apoptosis in comparison with that in SEV + LPS group. Similar results were demonstrated in Figure 2(B), LPS caused a decrease in Bcl2 expression, but increases in Bax, cleaved PARP, and cleaved caspase 3 expressions, indicating the induction of HK-2 apoptosis upon LPS stimulation. Nevertheless, the presence of SEV significantly recovered the expressions of these proteins altered by LPS, and EX527 co-treatment reversed the effects of SEV on LPS-induced HK-2 cells. The above results indicated that SEV could inhibit LPS-stimulated apoptosis of HK-2 cells and regulate apoptosis-related genes.
Figure 2 SEV inhibits LPS-induced apoptosis in HK-2 cells through activating SIRT1 expression. (A) TUNEL staining was utilized to measure HK-2 cell apoptosis. (B) The protein expressions of Bcl2, Bax, cleaved PARP, and cleaved caspase 3 were measured by western blot. N=3. ***P<0.001 vs. control; ###P<0.001 vs. LPS; ∆∆∆P<0.001 vs. SEV+LPS.
SEV protects HK-2 cells against LPS-induced inflammation and oxidative stress, whereas SIRT1 inhibition blocks this effect
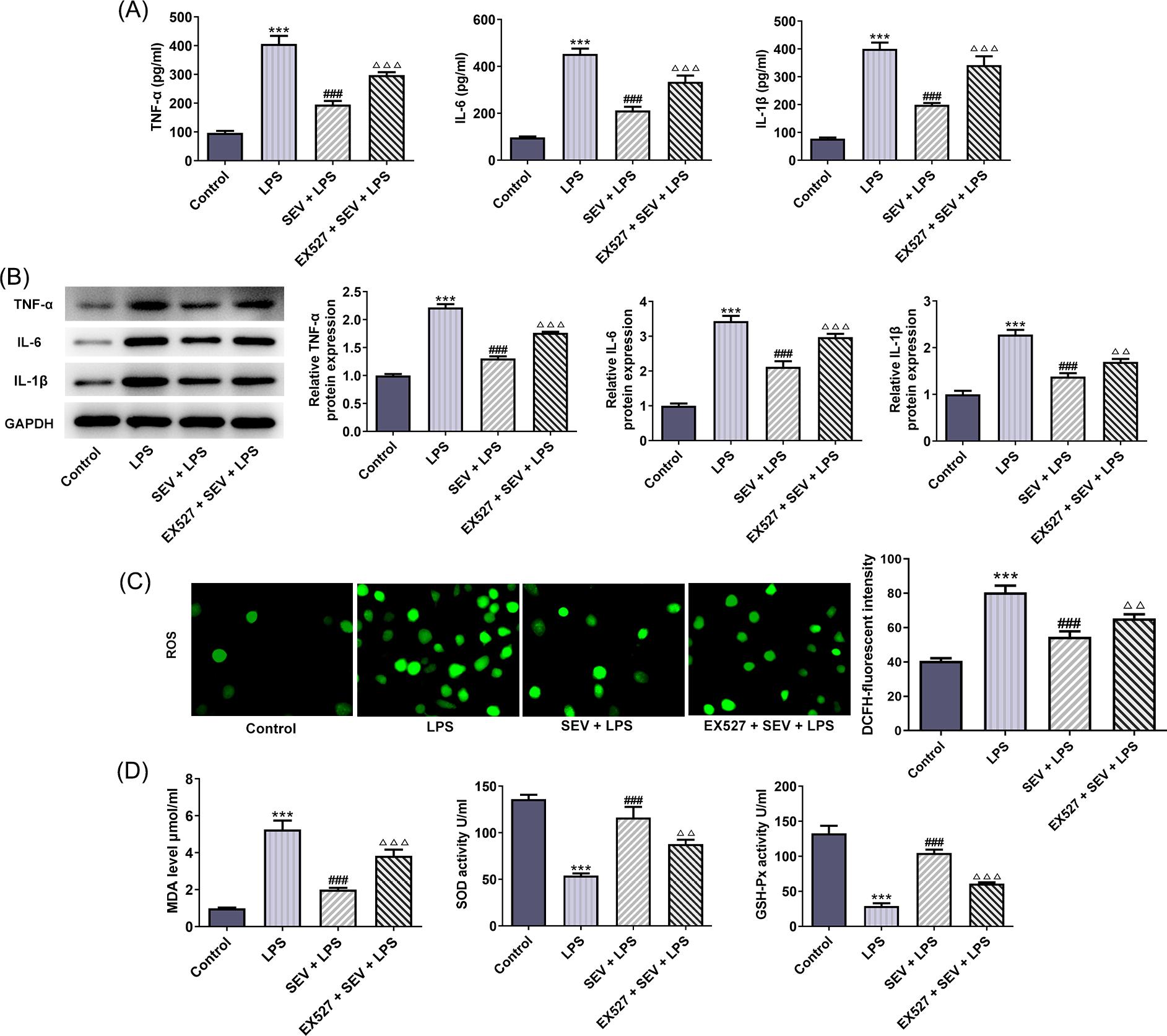
Subsequently, the changes in inflammation and oxidative stress were assessed. ELISA results from Figure 3(A) revealed that LPS stimulation remarkably enhanced the release of pro-inflammatory factors including TNF-α, IL-6, and IL-1β, but SEV pre-treatment significantly inhibited this effect. Meanwhile, EX527 co-treatment reversed the inhibitory effects of SEV on the production of pro-inflammatory cytokines induced by LPS in HK-2 cells. The same results were shown in Figure 3(B), which revealed that SEV down-regulated the protein expressions of TNF-α, IL-6, and IL-1β in LPS-treated HK-2 cells, whereas EX527 blocked this effect. LPS also led to a marked enhancement in cellular ROS, MDA levels, and SOD activity, but a reduction in GSH-Px activity (Figure 3C–D). However, SEV pre-treatment reduced ROS, MDA levels, and SOD activity and enhanced GSH-Px activity compared with that in cells with LPS treatment. EX527 co-treatment further reversed the effects of SEV. The above data illustrated that SEV could inhibit LPS-caused inflammation and oxidative stress in HK-2 cells.
Figure 3 SEV suppresses LPS-induced inflammation and oxidative stress in HK-2 cells through activating SIRT1 expression. (A) ELISA was employed to determine the production of TNF-α, IL-6, and IL-1β. (B) The protein expressions of TNF-α, IL-6, and IL-1β were measured by western blot. (C) ROS production was observed via DCFDA Cellular ROS Kit. (D) The levels of MDA, SOD, and GSH-Px were determined by corresponding kits. N=3. ***P<0.001 vs. control; ###P<0.001 vs. LPS; ∆∆P<0.01 and ∆∆∆P<0.001 vs. SEV+LPS.
SEV inhibits the activation of NF-κB signaling induced by LPS in HK-2 cells via up-regulating SIRT1 expression
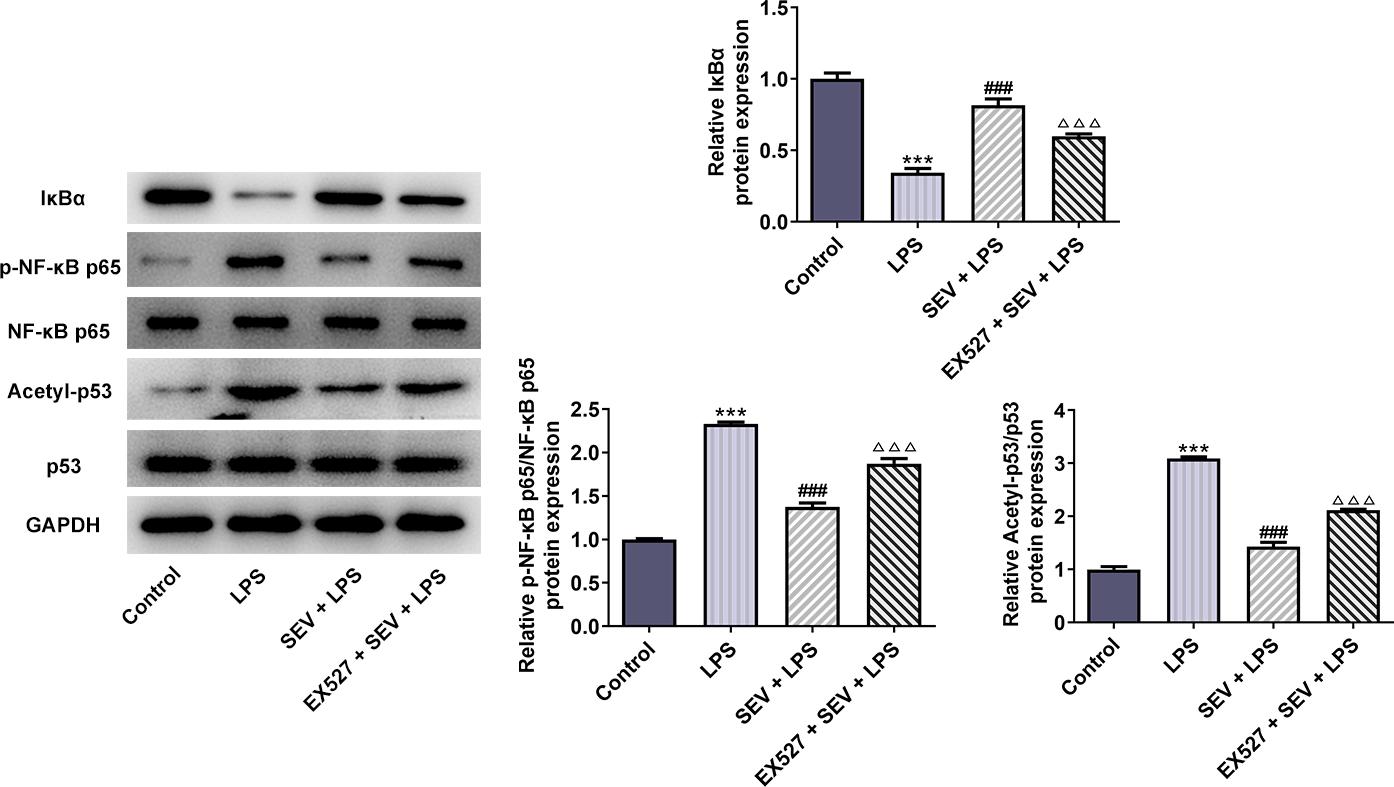
Finally, the expressions of SIRT1 and NF-κB signaling were detected. As shown in Figure 4, the expressions of SIRT1 and IκBα in HK-2 cells were down-regulated, while the contents of p-p65 and acetyl-p53 were up-regulated by LPS, indicating the activation of NF-κB signaling. In addition, the presence of SEV reversed the above phenomenon caused by LPS. Meanwhile, the suppressive effects of SEV on NF-κB signaling activation and the up-regulation of SIRT1 expression caused by SEV in LPS-treated HK-2 cells were also abolished by EX527.
Figure 4 SEV suppresses LPS-induced NF-κB activation in HK-2 cells through activating SIRT1 expression. The protein expressions of SIRT1, IκBα, p-NF-κB p65/ NF-κB p65, and acetyl-p53/p53 in HK-2 cells in different groups were measured by western blot. N=3. ***P<0.001 vs. control; ###P<0.001 vs. LPS; ∆∆P<0.01 and ∆∆∆P<0.001 vs. SEV+LPS.
Discussion
Sevoflurane is a well-known anesthetic inhalation gas and is extensively applied in general surgery. The increasing number of studies have indicated that SEV treatment protected organs such as the brain,5,6 lung,7 heart,8 and liver9 from sepsis-induced injury. In this study, we demonstrated that SEV pretreatment exerted significantly protective effects on HK-2 cells against LPS-induced inflammation, apoptosis, and oxidative stress along with activation of the NF-κB pathway.
LPS is a major component of the outer membrane of Gram-negative bacteria, the infection of which is one of the dominant causes of sepsis.26 The human renal tubular epithelial cells were the predominant cells affected by LPS-caused kidney injury. Therefore, LPS-induced HK-2 was chosen as the SI-AKI cell model. Following previous studies, we found that LPS remarkably caused acute inflammation, increased apoptosis, and oxidative stress in HK-2 cells.27–29 Tubular cell inflammation, apoptosis, and oxidative stress have been considered the major histopathological changes of SI-AKI. These results indicated the successful establishment of the SI-AKI cell model.
Our results showed that LPS-caused cell viability damage and LDH release were significantly reversed by SEV, suggesting the beneficial role of SEV in protecting HK-2 cells from LPS-induced cytotoxicity. Further experiments demonstrated that SEV suppressed LPS-induced apoptosis and inflammation in HK-2 cells. The inhibitory effects of SEV on apoptosis and inflammation have been reported in both in vivo and in vitro experiments.30 For example, SEV was found to reduce neutrophil apoptosis.31 In a vulnerable arterial plaque animal model, SEV promoted plaque stabilization and suppressed plaque disruption by enhancing collagen deposition and inhibiting inflammation.32 SEV also caused decreased levels of ROS and MDA, as well as increased activities of SOD and GSH-Px, in LPS-treated HK-2 cells. The intracellular antioxidant enzymes such as SOD and GSH-Px are responsible for eliminating the over-production of ROS and lipid peroxidation end product MDA. It is known that during AKI, superoxide production is increased and antioxidant enzyme activity is inhibited.29 Our results for the first time implicated the inhibitory effects of SEV on apoptosis, inflammation, and oxidative stress in SI-AKI.
The NF-κB signaling has been largely reported to play a key role in the pathogenesis of organ injury induced by sepsis.33,34 Inhibition of the NF-κB pathway has been confirmed to be associated with anti-inflammatory activity, thus alleviating SI-AKI.35 Our results showed that SEV up-regulated IκBα expression and down-regulated the expressions of p-p65 and acetyl-p53 in LPS-induced HK-2 cells, indicating that SEV could inhibit NF-κB pathway activation caused by LPS.
Moreover, LPS-induced down-regulation of SIRT1 and up-regulation of acetyl-p53 were also reversed by SEV. Sirtuins are histone deacetylases, and play a significant role in inflammation, energy metabolism, oxidative stress, and cancer.36,37 SIRT1 could deacetylate p53, and it was found that the deacetylation of p53 caused by SIRT1 could protect cells from p53-dependent apoptosis or senescence.38 SIRT1 is involved in the process of acute kidney injury, and it can inhibit the apoptosis of renal tubular epithelial cells induced by hypoxia and reoxygenation, TGF-β1 and high glucose, as well as resist renal tissue fibrosis.39–42 Studies have shown that SIRT1 also has an anti-inflammatory function, which has been proven in vascular tissue inflammation, airway inflammation, and spinal cord inflammation.43 From this, SIRT1 can regulate inflammation, oxidative stress, and apoptosis of renal tubular epithelial cells.
A previous study demonstrated that SEV up-regulated SIRT1 expression in a sepsis model.6 Therefore, we speculated that SEV may exert its protective effects against LPS-induced HK-2 injury by increasing SIRT1 expression, thereby inhibiting NF-κB activation and promoting p53 deacetylation. To validate this hypothesis, an inhibitor of SIRT1, EX527 was employed to challenge LPS and SEV-treated HK-2 cells. Scientists from Elixir Pharmaceuticals first discovered indol EX527, the first selective SIRT1 inhibitor, in 2005.44 The molecular structure of EX527 has a carboxyamide part, which is similar to the acyl part of nicotinamide. Blocking the formation of 2-O-acetyl-ADP ribose and SIRT1 complex is the mechanism of EX527 inhibiting SIRT1.45 Consistent with the above speculation, the presence of EX527 significantly blunted all effects of SEV on HK-2 cells, indicating the indispensable role of SIRT1 expression in the actions of SEV.
This study presented that SEV could regulate the SIRT1 expression. Previous studies indicated that the apoptosis and inflammatory response of renal tubular epithelial cells was enhanced by suppressing the SIRT1 expression and activating the NF-κB signaling pathway46 and sepsis-induced inflammation and apoptosis could be alleviated by inhibiting the NF-κB pathway via targeting SIRT1.47 Therefore, we speculated that SEV suppressed the apoptosis of LPS-induced HK-2 cells by promoting the SIRT1 expression and inactivating the NF-κB signaling pathway.
Conclusions
Taken together, the present study revealed that SEV could alleviate sepsis-induced kidney injury in vitro via inhibiting renal tubular epithelial cells apoptosis, inflammation, and oxidative stress. Furthermore, mitigative effects of SEV on SI-AKI were mainly through up-regulating SIRT1 expression to suppress NF-κB pathway activation. SEV might be a potential clinical therapeutic choice for SI-AKI. However, the present study just demonstrates the protective role of SEV in the damage of kidney cells and the present findings can’t translate into a clinical situation. Further animal experiments and clinical trials need to be conducted to verify the effects of SEV. The potential negative effects of SEV on kidney function should also be explored. In addition, the mechanism by which SEV promotes SIRT1 expression will be explored in our future study.
Data availability
The data included in this study can be available from the corresponding author upon reasonable request.
Funding statement
This work was supported by The Science and Technology Project (Social Development) of Wujin District [grant No. WS202008].
REFERENCES
1. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96:1083–1099. 10.1016/j.kint.2019.05.026
2. Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ (Clinical research ed). 2019;364:k4891. 10.1136/bmj.k4891
3. Levey AS, James MT. Acute kidney injury. Ann Intern Med. 2017;167:Itc66-itc80. 10.7326/AITC201711070
4. Rahman M, Shad F, Smith MC. Acute kidney injury: A guide to diagnosis and management. Am Fam Physician. 2012;86:631–639. PMid: 23062091
5. Bedirli N, Bagriacik EU, Yilmaz G, Ozkose Z, Kavutçu M, Cavunt Bayraktar A, et al. Sevoflurane exerts brain-protective effects against sepsis-associated encephalopathy and memory impairment through caspase 3/9 and Bax/Bcl signaling pathway in a rat model of sepsis. J Int Med Res. 2018;46:2828–2842. 10.1177/0300060518773265
6. Chen H, Peng Y, Wang L, Wang X. Sevoflurane attenuates cognitive dysfunction and NLRP3-dependent caspase-1/11-GSDMD pathway-mediated pyroptosis in the hippocampus via upregulation of SIRT1 in a sepsis model. Arch Physiol Biochem. 2020;??:1–8. 10.1080/13813455.2020.1773860
7. Zhang E, Zhao X, Ma H, Luo D, Hu Y, Hou L, et al. A subanesthetic dose of sevoflurane combined with oxygen exerts bactericidal effects and prevents lung injury through the nitric oxide pathway during sepsis. Biomed Pharmacother. 2020;127:110169. 10.1016/j.biopha.2020.110169
8. Li J, Liu P, Li H, Wang Y, Chen Y, Qi R, Sevoflurane preconditioning prevents septic myocardial dysfunction in lipopolysaccharide-challenged mice. J Cardiovasc Pharmacol. 2019;74:462–473. 10.1097/FJC.0000000000000734
9. Xiao X, Liu D, Chen S, Li X, Ge M, Huang W. Sevoflurane preconditioning activates HGF/Met-mediated autophagy to attenuate hepatic ischemia-reperfusion injury in mice. Cell Signal. 2021;82:109966. 10.1016/j.cellsig.2021.109966
10. Rodríguez-González R, Baluja A, Veiras Del Río S, Rodríguez A, Rodríguez J, Taboada M, et al. Effects of sevoflurane postconditioning on cell death, inflammation and TLR expression in human endothelial cells exposed to LPS. J Transl Med. 2013;11:87. 10.1186/1479-5876-11-87
11. Li H, Weng Y, Yuan S, Liu W, Yu H, Yu W. Effect of sevoflurane and propofol on acute kidney injury in pediatric living donor liver transplantation. Ann Transl Med. 2019;7:340. 10.21037/atm.2019.06.76
12. Bonkowski MS, Sinclair DA. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17:679–690. 10.1038/nrm.2016.93
13. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood. 2019;133:266–279. 10.1182/blood-2018-07-863233
14. Hu X, Lu Z, Yu S, Reilly J, Liu F, Jia D, et al. CERKL regulates autophagy via the NAD-dependent deacetylase SIRT1. Autophagy. 2019;15:453–465. 10.1080/15548627.2018.1520548
15. Zhu S, Dong Z, Ke X, Hou J, Zhao E, Zhang K, et al. The roles of sirtuins family in cell metabolism during tumor development. Semin Cancer Biol. 2019;57:59–71. 10.1016/j.semcancer.2018.11.003
16. Wei S, Gao Y, Dai X, Fu W, Cai S, Fang H, et al. SIRT1-mediated HMGB1 deacetylation suppresses sepsis-associated acute kidney injury. Am J Physiol Renal Physiol. 2019;316: F20–F31. 10.1152/ajprenal.00119.2018
17. Cheng X, Zhang S, Wen Y, Shi Z. Clinical significance of sirtuin 1 level in sepsis: correlation with disease risk, severity, and mortality risk. Braz J Med Biol Res. 2020;54:e10271. 10.1590/1414-431x202010271
18. Ma Y, Nie H, Chen H, Li J, Hong Y, Wang B, et al. NAD+/NADH metabolism and NAD+-dependent enzymes in cell death and ischemic brain injury: Current advances and therapeutic implications. Curr Med Chem. 2015;22:1239–1247. 10.2174/0929867322666150209154420
19. Kim DH, Jung YJ, Lee JE, Lee AS, Kang KP, Lee S, et al. SIRT1 activation by resveratrol ameliorates cisplatin-induced renal injury through deacetylation of p53. Am J Physiol Renal Physiol. 2011;301:F427–F435. 10.1152/ajprenal.00258.2010
20. Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2013;83:404–413. 10.1038/ki.2012.394
21. Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. JASN. 2011;22:1505–1516. 10.1681/ASN.2011020203
22. Huang G, Hao F, Hu X. Downregulation of microRNA-155 stimulates sevoflurane-mediated cardioprotection against myocardial ischemia/reperfusion injury by binding to SIRT1 in mice. J Cell Biochem. 2019;120:15494–15505. 10.1681/ASN.2011020203
23. Shi C, Zhao Y, Li Q, Li J. lncRNA SNHG14 plays a role in sepsis-induced acute kidney injury by regulating miR-93. Mediat Inflamm. 2021;2021:5318369. 10.1155/2021/5318369
24. Hou T, Ma H, Wang H, Chen C, Ye J, Ahmed AM, et al. Sevoflurane preconditioning attenuates hypoxia/reoxygenation injury of H9c2 cardiomyocytes by activation of the HIF-1/PDK-1 pathway. PeerJ. 2020;8:e10603. 10.7717/peerj.10603
25. Liu ZH, Zhang Y, Wang X, Fan XF, Zhang Y, Li X, et al. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed Pharmacother. 2019;118:109227. 10.1016/j.biopha.2019.109227
26. Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, et al. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci USA. 2009;106:2348–2352. 10.1073/pnas.0808146106
27. Zhang Y, Wang L, Meng L, Cao G, Wu Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle (Georgetown, Tex). 2019;18:425–436. 10.1080/15384101.2019.1568746
28. Huang G, Bao J, Shao X, Zhou W, Wu B, Ni Z, et al. Inhibiting pannexin-1 alleviates sepsis-induced acute kidney injury via decreasing NLRP3 inflammasome activation and cell apoptosis. Life Sci. 220;254:117791. 10.1016/j.lfs.2020.117791
29. Wang Z, Wu J, Hu Z, Luo C, Wang P, Zhang Y, et al. Dexmedetomidine alleviates lipopolysaccharide-induced acute kidney injury by inhibiting p75NTR-mediated oxidative stress and apoptosis. Oxid Med Cell Longev. 2020;2020:5454210. 10.1016/j.lfs.2020.117791
30. Ngamsri KC, Fabian F, Fuhr A, Gamper-Tsigaras J, Straub A, Fecher D, et al. Sevoflurane exerts protective effects in murine peritonitis-induced sepsis via hypoxia-inducible factor 1α/adenosine A2B receptor signaling. Anesthesiology. 2021;135:136–150. 10.1097/ALN.0000000000003788
31. Koutsogiannaki S, Hou L, Babazada H, Okuno T, Blazon-Brown N, Soriano SG, et al. The volatile anesthetic sevoflurane reduces neutrophil apoptosis via Fas death domain-Fas-associated death domain interaction. FASEB J. 2019;33:12668–12679. 10.1096/fj.201901360R
32. Hou Y, Lin X, Lei Z, Zhao M, Li S, Zhang M, et al. Sevoflurane prevents vulnerable plaque disruption in apolipoprotein E-knockout mice by increasing collagen deposition and inhibiting inflammation. Br J Anaesth. 2020;125:1034–1044. 10.1016/j.bja.2020.07.054
33. Brown MA, Jones WK. NF-kappaB action in sepsis: The innate immune system and the heart. Front Biosci. 2004;9:1201–1217. 10.2741/1304
34. Liu Z, Tang C, He L, Yang D, Cai J, Zhu J, et al. The negative feedback loop of NF-κB/miR-376b/NFKBIZ in septic acute kidney injury. JCI Insight. 2020;5: e142272. 10.1172/jci.insight.142272
35. Li YM, Zhang J, Su LJ, Kellum JA, Peng ZY. Downregulation of TIMP2 attenuates sepsis-induced AKI through the NF-κb pathway. Biochim Biophys Acta-Mol Basis Dis. 2019;1865:558–569. 10.1172/jci.insight.142272
36. Tang XL, Wang X, Fang G, Zhao YL, Yan J, Zhou Z, et al. Resveratrol ameliorates sevoflurane-induced cognitive impairment by activating the SIRT1/NF-κB pathway in neonatal mice. J Nutr Biochem. 2021;90:108579. 10.1016/j.jnutbio.2020.108579
37. Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, et al. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J Hepatol. 2017;67:1232–1242. 10.1016/j.jhep.2017.08.010
38. Ong ALC, Ramasamy TS. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Age Res Rev. 2018;43:64–80. 10.1016/j.arr.2018.02.004
39. Zhou L, Xu DY, Sha WG, Shen L, Lu GY, Yin X, et al. High glucose induces renal tubular epithelial injury via Sirt1/NF-kappaB/microR-29/Keap1 signal pathway. J Transl Med. 2015;13:352. 10.1186/s12967-015-0710-y
40. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun. 2008;372:51–56. 10.1016/j.bbrc.2008.04.176
41. Xu S, Zeng Z, Zhao M, Huang Q, Gao Y, Dai X, et al. Evidence for SIRT1 mediated HMGB1 release from kidney cells in the early stages of hemorrhagic shock. Front Physiol. 2019;10:854. 10.3389/fphys.2019.00854
42. Li P, Liu Y, Qin X, Chen K, Wang R, Yuan L, et al. SIRT1 attenuates renal fibrosis by repressing HIF-2α. Cell Death Discov. 2021;7:59. 10.1038/s41420-021-00443-x
43. Hwang JW, Yao H, Caito S, Sundar IK, Rahman I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med. 2013;61:95–110. 10.1016/j.freeradbiomed.2013.03.015
44. Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045–8054. 10.1021/jm050522v
45. Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci USA. 2013;110:E2772–E2781. 10.1021/jm050522v
46. Han S, Lin F, Ruan Y, Zhao S, Yuan R, Ning J, et al. miR-132-3p promotes the cisplatin-induced apoptosis and inflammatory response of renal tubular epithelial cells by targeting SIRT1 via the NF-κB pathway. Int Immunopharmacol.2021;99:108022. 10.1021/jm050522v
47. Wu Z, Chen J, Zhao W, Zhuo CH, Chen Q. Inhibition of miR-181a attenuates sepsis-induced inflammation and apoptosis by activating Nrf2 and inhibiting NF-κB pathways via targeting SIRT1. Kaohsiung J Med Sci. 2021;37:200–207. 10.1002/kjm2.12310
Figure S1 SEV has no side effect on the biological behavior of HK-2 cells. The viability of HK-2 cells treated by SEV was detected by CCK-8 assay. N=3.