
Download
ORIGINAL RESEARCH
Knockdown of CXCL3-inhibited apoptosis and inflammation in lipopolysaccharide-treated BEAS-2B and HPAEC through inactivating MAPKs pathway
Yuhui Wanga, Linyan Panb*
aIntensive Care Unit, Guizhou Provincial People's Hospital, Guiyang, Guizhou Province, China
bDepartment of Respiratory Medicine, Wenzhou Central Hospital, Wenzhou, Zhejiang Province, China
Abstract
Background: CXCL3 (C-X-C motif chemokine ligand 3) is a member of chemokines family, which binds to the receptor to recruit neutrophils to lungs, thus participating in the pathogenesis of asthmatic lung. The role of CXCL3 in sepsis-induced acute lung injury is investigated here.
Methods: Human lung epithelial cell line (BEAS-2B) and human pulmonary artery endothelial cell line (HPAEC) were treated with lipopolysaccharides (LPS). MTT and flow cytometry were performed to detect cell viability and apoptosis, respectively. Enzyme-linked immunosorbent assay (ELISA) and real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) were used to assess the levels of inflammatory factors.
Results: Treatment with LPS resulted in the decrease of cell viability in BEAS-2B and HPAEC. CXCL3 was particularly upregulated in LPS-treated BEAS-2B and HPAE cells. Knockdown of CXCL3 enhanced viability and suppressed apoptosis i006E LPS-treated BEAS-2B and HPAE cells. Knockdown of CXCL3 also upregulated TNF-α, IL-1β, and IL-18 in LPS-treated BEAS-2B and HPAE cells. Moreover, knockdown of CXCL3 suppressed the activation of mitogen-activated protein kinases (MAPKs) signaling in LPS-treated BEAS-2B and HPAE cells through downregulation of p-ERK1/2, p-p38, and p-JNK. On the other hand, overexpression of CXCL3 caused completely opposite results in LPS-treated BEAS-2B and HPAE cells.
Conclusion: Knockdown of CXCL3 exerted antiapoptotic and anti-inflammatory effects against LPS-treated BEAS-2B and HPAE cells, at least partially, through inactivation of MAPKs signaling, suggesting a potential strategy for the intervention of sepsis-induced acute lung injury.
Key words: beas-2b, cxcl3, hpaec, inflammation, lipopolysaccharide, mapks, sepsis
*Corresponding author: Linyan Pan, Department of Respiratory Medicine, Wenzhou Central Hospital, No. 252 Baili East Road, Lucheng District, Wenzhou, Zhejiang Province, China. Email address: [email protected]
Received 3 March 2022; Accepted 22 March 2022; Available online 1 July 2022
Copyright: Wang Y and Pan L
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Sepsis, a systemic pathophysiological and biochemical disorder, is caused by severe bacterial, viral, parasitic, or fungal infection.1 Sepsis, as an inflammatory response syndrome, stimulates multiple organ failure.2 Sepsis and the related diseases contribute to approximately 20% of all deaths globally.3 Lung is one of the most susceptible organs to sepsis, and sepsis-induced acute lung injury is a common complication that accounts for high morbidity and mortality.4 However, effective therapies for sepsis-induced acute lung injury are currently devoid.
Lipopolysaccharide (LPS), an endotoxin found in cell wall of gram-negative bacteria, has been reported to induce excessive immune response in lungs and contribute to the pathogenesis of sepsis-induced acute lung injury.5 Amelioration of LPS-induced inflammation attenuated -sepsis-associated acute lung injury.6 TAK-242 functioned as an inhibitor of TLR4, and eritoran antagonized the binding of LPS to TLR4, thus blocking gram-negative bacterial infection–induced inflammatory responses in sepsis.7,8 However, clinical trials showed that antagonists of TLR4, TAK-242, and eritoran tetrasodium showed no optimistic results in septic patients.9–11 Therefore, other mechanisms involved in inflammation of sepsis-induced acute lung injury should be investigated.
Chemokines play a key role in innate and adaptive immunity through regulation in the polarization of CD4+ T cells, cell activation, microbicidal activity, and cell recruitment.12 Production of chemokines and inflammatory cytokines are involved in the pathogenesis of sepsis-associated acute lung injury.13 For example, CXC chemokine ligand 1 (CXCL1) regulated neutrophil migration and Th17 differentiation to facilitate the host defense in polymicrobial sepsis.14 CXCL3 is a member of chemokine family that mediates vascular remodeling and mobilization and trafficking of fibrocytes during the development of pulmonary fibrosis.15 CXCL3 also functions as a ligand for CXC chemokine receptor 2 (CXCR2), and CXCL3/CXCR2 axis regulates the infiltration of neutrophils to the inflammatory sites, and is therefore associated with inflammatory diseases.16 CXCL3 recruited CXCR2-positive neutrophils to promote rhinoviral-induced inflammation during the development of asthma exacerbation.17 Moreover, CXCL3 was found to be a differentially expressed gene in mechanical ventilation and LPS-induced mice with acute lung injury.18 However, the role of CXCL3 in sepsis-associated acute lung injury has not been reported yet.
In this study, the effects of CXCL3 on cell apoptosis and inflammation of LPS-treated human lung epithelial cell line (BEAS-2B) and human pulmonary artery endothelial cell line (HPAEC) were investigated, and the related mechanism was revealed. This study might provide potential strategy for the prevention of sepsis-associated acute lung injury.
Materials and Methods
Cell culture and treatment
BEAS-2B and HPAE cells were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA). Cells were incubated with 1, 5, or 10 μg/mL LPS (Sigma-Aldrich) for 24 h before the functional assays.
Cell viability and apoptosis assays
Full length of CXCL3 was subcloned into pcDNA-3.1 vector (Invitrogen, Carlsbad, CA, USA) to generate pc-CXCL3. siRNA targeting CXCL3 (siCXCL3) was synthesized by GenePharma (Suzhou, China). BEAS-2B and HPAE cells were seeded into a 96-well plate and grown in 5 μg/mL LPS for 24 h after transfection with 200 μg pcDNAs (pc-CXCL3 or NC) or 20 nM siRNAs (siCXCL3 or siNC). Cells were then treated with MTT solution (Beyotime, Beijing, China) for 4 h. The supernatant was removed, and formazan was dissolved with dimethyl sulfoxide (Sigma-Aldrich). Absorbance at 570 nm was measured using microplate reader (Thermo Fisher Scientific) to detect cell viability. For flow cytometry, LPS-treated (5 μg/mL) BEAS-2B and HPAE cell lines were transfected with pcDNAs or siRNAs. Cells were subjected to trypsin digestion, and then resuspended in binding buffer of BD CycletestTM Plus DNA Reagent Kit (BD Biosciences, San Jose, CA). Cells were stained with 5 μL fluorescein isothiocyanate–conjugated annexin V and 5 μL propidium oxide (1 mg/mL), and then analyzed under fluorescence activated cell sorting (FACS) flow cytometer (Life Technologies, Darmstadt, Germany).
qRT-PCR and ELISA
BEAS-2B and HPAE cells were lysed in TRIzol kit (Invitrogen, Carlsbad, CA, USA), and the isolated RNAs were synthesized into cDNAs using MultiscribeTM Reverse transcription Kit (Applied Biosystems, CA, USA). The mRNA expression of TNF-α, IL-1β, and IL-18 were detected by PreTaq II kit (Takara, Dalian, Liaoning, China) with the following primers: TNF-α (Forward: 5’-ATGGGCTCCCTCTCATCAGT-3’ and Reverse: 5’-GCTTGGTGGTTTGCTACGAC-3’); IL-1β (Forward: 5’-ATGAGGACCCAAGCACCTTC-3’ and Reverse: 5’-ACCACTT GTTGGCTTATGTTCTG-3’); IL-18 (Forward: 5’-AAAGTGCCA GTGAACCC-3’ and Reverse: 5’-TTTGATGTAAGTTAGTGAGA GTGA-3’). β-actin (Forward: 5’-TACTGCCCTGGCTCCTAGCA-3’ and Reverse: 5’-TGGACAGTGAGGCCAGGATAG-3’) was used as the internal control. For ELISA, the cultured medium of BEAS-2B and HPAE cells were harvested, and then subjected to the commercial ELISA kits (Thermo Fisher Scientific) to detect levels of TNF-α, IL-1β, and IL-18.
Western blot
BEAS-2B and HPAE cells were lysed in Radioimmunoprecipitation assay (RIPA) buffer (Beyotime), and the isolated proteins were then separated by 10% SDS-PAGE. Proteins were transferred onto nitrocellulose membranes, and the membranes were blocked in 5% bovine serum albumin. Membranes were then probed with specific antibodies: anti-CXCL3 and anti-β-actin (1:2000), anti-GAPDH (1:2500), anti-p-ERK1/2 and anti-ERK1/2 (1:3000), anti-p-JNK and anti-JNK (1:3500), anti-p-p38 and anti-p38 (1:4000). The membranes were then washed and incubated with horseradish peroxidase–conjugated secondary antibody (1:5000). Immunoreactivities were visualized using enhanced chemiluminescence (Sigma-Aldrich). All the antibodies were acquired from Abcam (Cambridge, MA, USA).
Statistical analysis
All data with at least triple replicates were expressed as mean ± SEM and analyzed by Student’s t-test or one-way analysis of variance under SPSS software. P ˂ 0.05 was considered as statistically significant.
Results
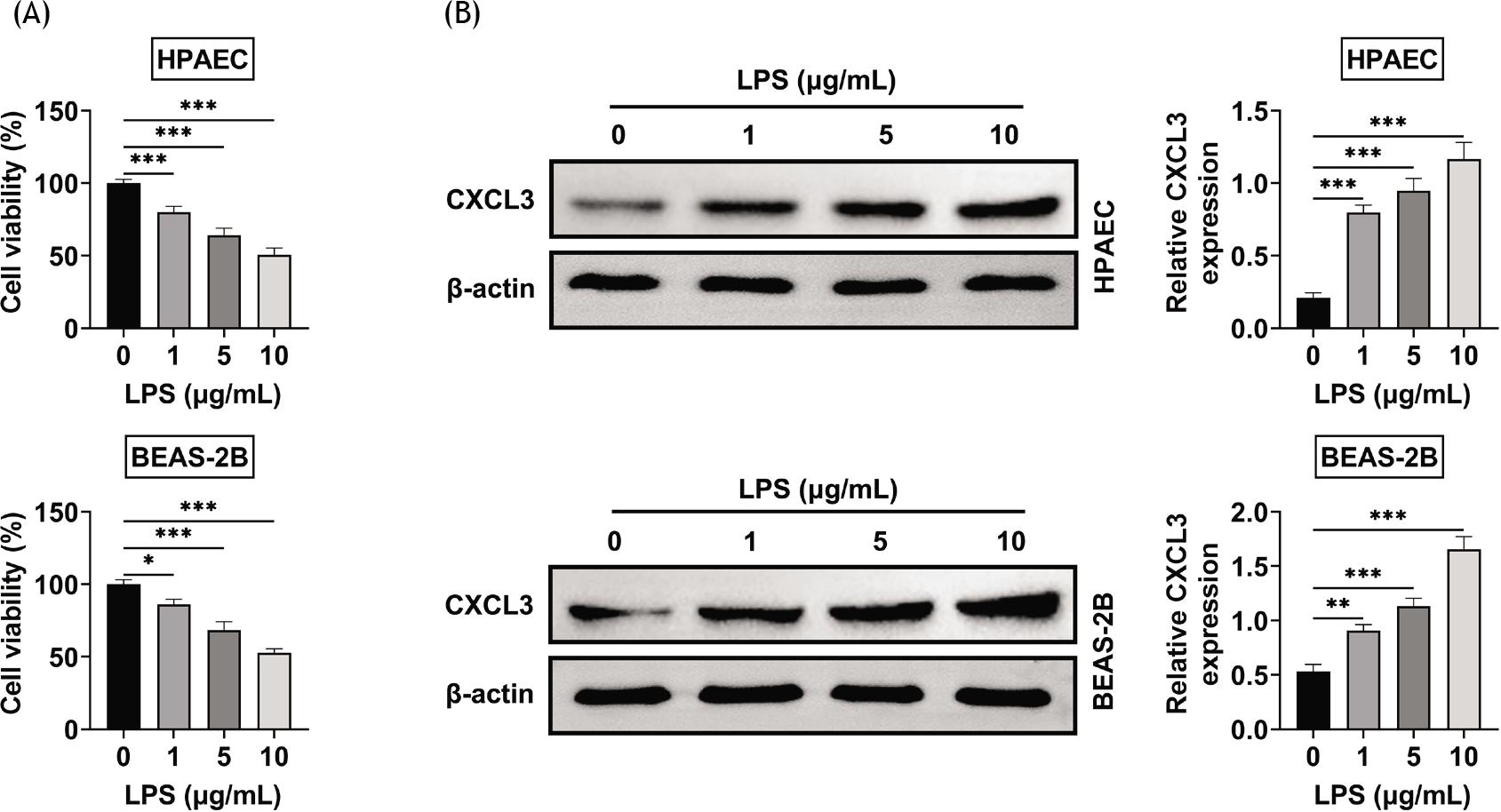
LPS caused the upregulation of CXCL3 in LPS-treated BEAS-2B and HPAE cells to induce cytotoxicity. The viability of BEAS-2B and HPAE cells were reduced by LPS in a dose-dependent manner (Figure 1A). Protein expression of CXCL3 in BEAS-2B and HPAE cells was enhanced by LPS in a dose-dependent manner (Figure 1B), which suggested the potential functional role of CXCL3 in LPS-treated BEAS-2B and HPAE cells.
Figure 1 Lipopolysaccharides caused the upregulation of CXCL3 in BEAS-2B and HPAE cells. (A) The viabilities of BEAS-2B and HPAE cells were reduced by lipopolysaccharides in a dose-dependent manner. (B) The protein expression of CXCL3 in BEAS-2B and HPAE cells was enhanced by lipopolysaccharides in a dose-dependent manner. *, **, ***, P ˂ 0.05, P ˂ 0.01, P ˂ 0.001. BEAS-2B, human lung epithelial cells; CXCL3, C-X-C motif chemokine ligand 3; HPAEC, human pulmonary artery endothelial cells.
Knockdown of CXCL3 inhibited the apoptosis of LPS-treated BEAS-2B and HPAEC
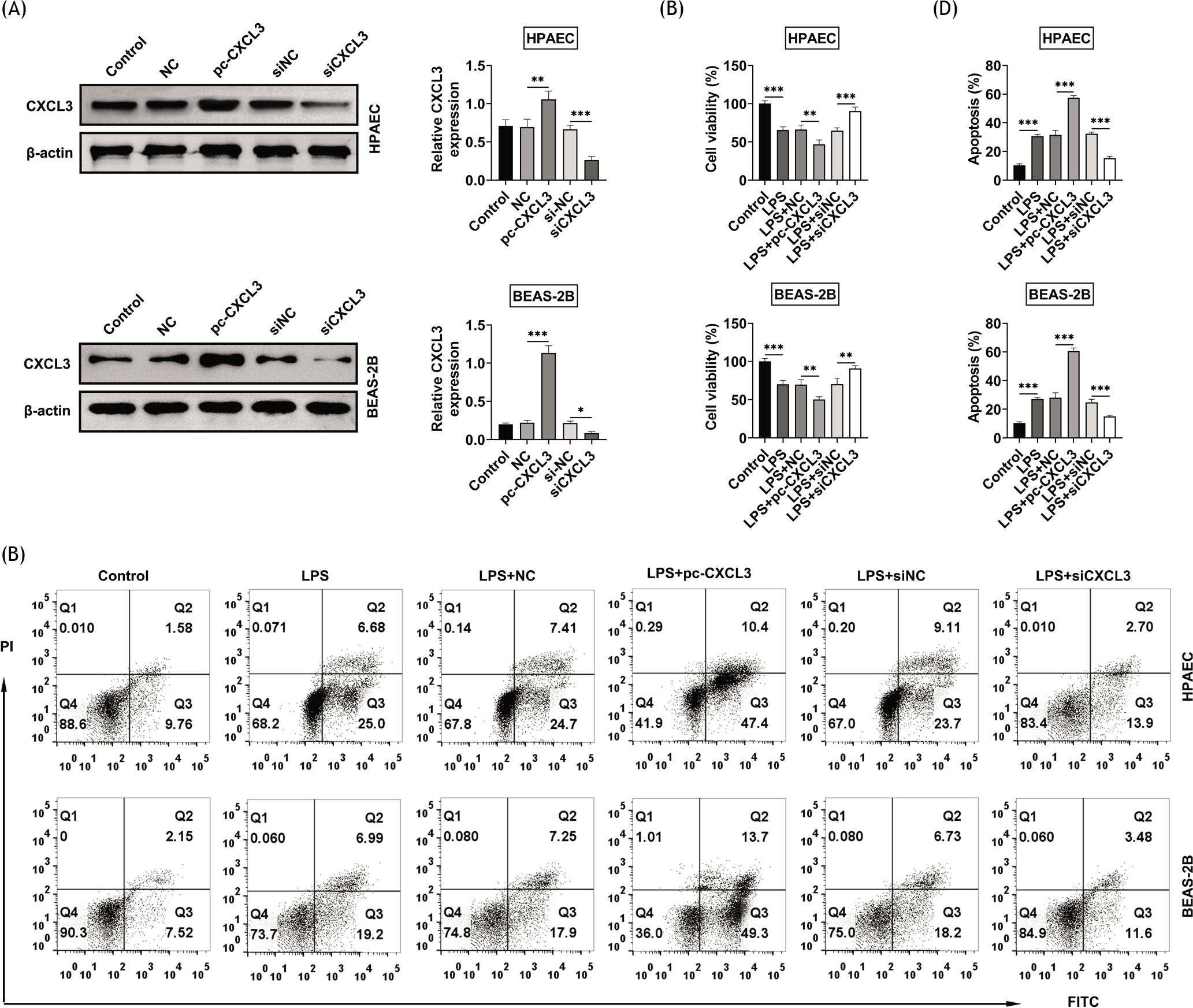
LPS-treated BEAS-2B and HPAE cell lines were then subjected to gain- and loss- of functional assays through transfection with pcDNAs and siRNAs, respectively. Transfection with pc-CXCL3 or siCXCL3 increased or decreased the protein expression of CXCL3 in BEAS-2B and HPAE cells, respectively (Figure 2A). Overexpression of CXCL3 reduced the viabilities, while knockdown of CXCL3 enhanced the viabilities in LPS-treated BEAS-2B and HPAE cell lines (Figure 2B). Moreover, apoptosis of LPS-treated BEAS-2B and HPAEC was promoted by overexpression of CXCL3, while the process was inhibited by knockdown of CXCL3 (Figures 2C and 2D), demonstrating the antiapoptotic effect of CXCL3 silence against LPS-treated BEAS-2B and HPAEC cells.
Figure 2 Knockdown of CXCL3 inhibited the apoptosis of LPS-treated BEAS-2B and HPAE cell lines. (A) Transfection with pc-CXCL3 or siCXCL3 increased or decreased the protein expression of CXCL3 in BEAS-2B and HPAE cells, respectively. (B) Overexpression of CXCL3 reduced the viabilities of LPS-treated BEAS-2B and HPAE cell lines, while knockdown of CXCL3 caused the opposite results. (C) Overexpression of CXCL3 promoted the apoptosis of LPS-treated BEAS-2B and HPAE cell lines, while knockdown of CXCL3 caused the opposite results. (D) Relative apoptosis of LPS-treated BEAS-2B and HPAE cell lines after transfection of pc-CXCL3 or siCXCL3, respectively. *, **, ***, P ˂ 0.05, P ˂ 0.01, P ˂ 0.001. BEAS-2B, human lung epithelial cells; CXCL3, C-X-C motif chemokine ligand 3; HPAEC, human pulmonary artery endothelial cells; LPS, lipopolysaccharides.
Knockdown of CXCL3 inhibited the inflammation of LPS-treated BEAS-2B and HPAE cell lines
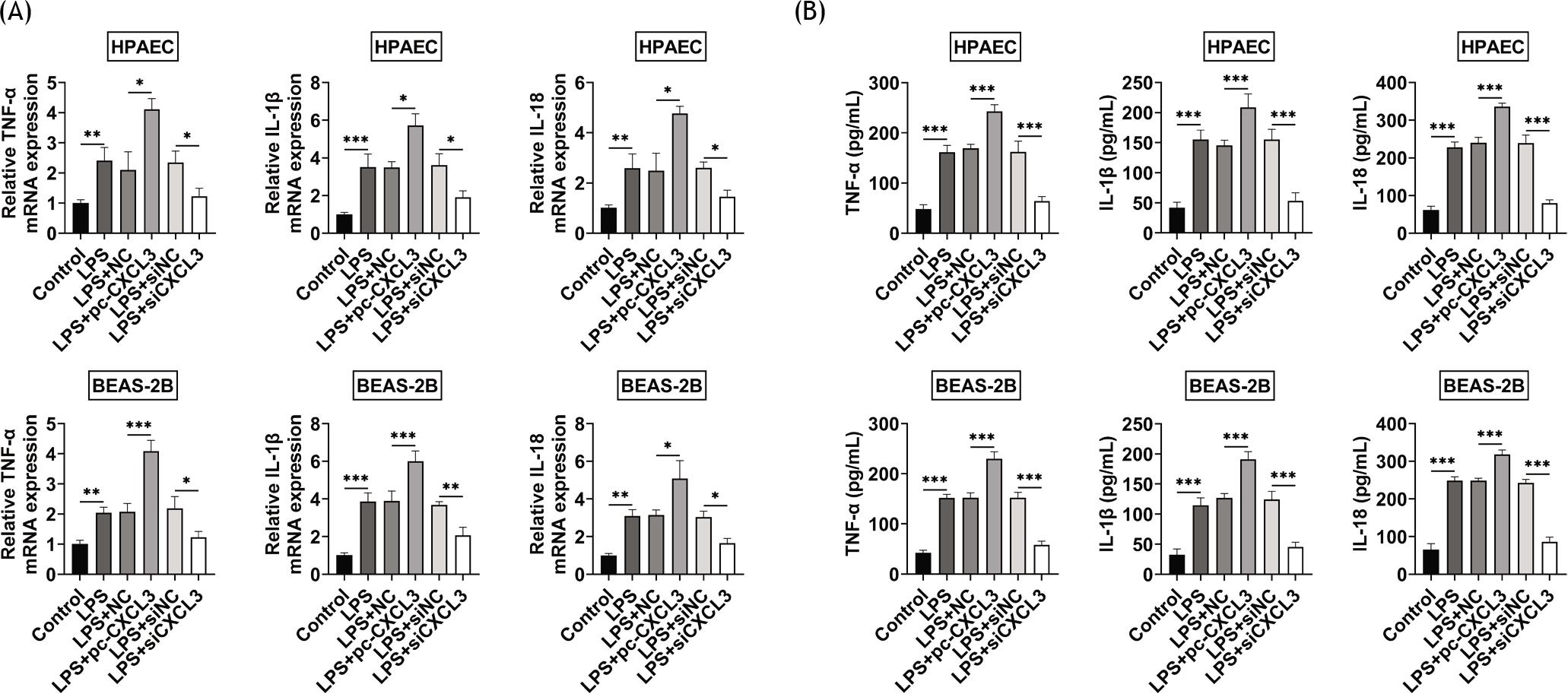
LPS induced the upregulation of TNF-α, IL-1β, and IL-18 in BEAS-2B and HPAE cells (Figures 3A and 3B). Overexpression of CXCL3 upregulated the levels of TNF-α, IL-1β, and IL-18, while knockdown of CXCL3 downregulated the levels of these cytokines in LPS-treated BEAS-2B and HPAE cells (Figures 3A and 3B), exerting the anti-inflammatory effect of CXCL3 silence against LPS-treated BEAS-2B and HPAE cells.
Figure 3 Knockdown of CXCL3 inhibited the inflammation of LPS-treated BEAS-2B and HPAE cell lines. (A) Overexpression of CXCL3 upregulated the mRNA expressions of TNF-α, IL-1β, and IL-18, while knockdown of CXCL3 downregulated the levels of TNF-α, IL-1β, and IL-18 in LPS-treated BEAS-2B and HPAE cells. (B) Overexpression of CXCL3 upregulated the protein expressions of TNF-α, IL-1β, and IL-18, while knockdown of CXCL3 down-regulated the levels of TNF-α, IL-1β, and IL-18 in LPS-treated BEAS-2B and HPAE cells. *, **, ***, P ˂ 0.05, P ˂ 0.01, P ˂ 0.001. BEAS-2B, human lung epithelial cells; CXCL3, C-X-C motif chemokine ligand 3; HPAEC, human pulmonary artery endothelial cells; LPS, lipopolysaccharides.
Knockdown of CXCL3-inhibited MAPKs signaling in LPS-treated BEAS-2B and HPAE cell lines
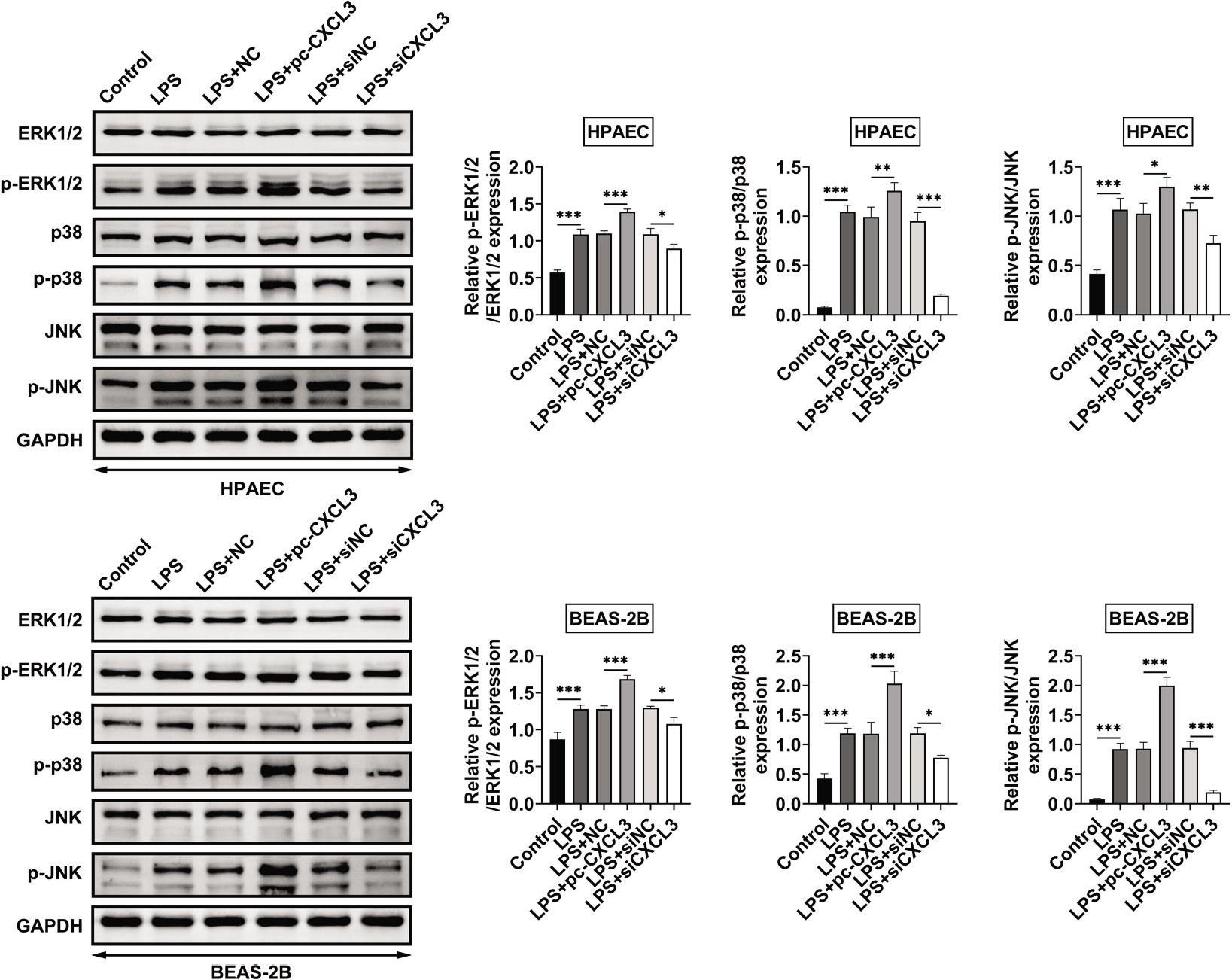
The protein expressions of p-ERK1/2, p-p38, and p-JNK in BEAS-2B and HPAE cells were increased by LPS (Figure 4). Overexpression of CXCL3 promoted the activation, while knockdown of CXCL3 suppressed the activation of MAPKs signaling in LPS-treated BEAS-2B and HPAE cells through upregulation and downregulation of p-ERK1/2, p-p38, and p-JNK, respectively (Figure 4).
Figure 4 Knockdown of CXCL3 inhibited MAPKs signaling in LPS-treated BEAS-2B and HPAEC. Overexpression promoted the upregulation of p-ERK1/2, p-p38, and p-JNK, while knockdown of CXCL3 reduced the levels of p-ERK1/2, p-p38, and p-JNK in LPS-treated BEAS-2B and HPAE cells. *, **, ***, P ˂ 0.05, P ˂ 0.01, P ˂ 0.001. BEAS-2B, human lung epithelial cells; CXCL3, C-X-C motif chemokine ligand 3; HPAEC, human pulmonary artery endothelial cells; LPS, lipopolysaccharides; MAPKs, mitogen-activated protein kinases.
Discussion
Inflammatory characteristics of LPS-induced acute lung injury in mice showed that CXCLs were primarily accumulated in lung tissues.19 LPS induced upregulation of CXCL4 and contributed to apoptosis and permeability of endothelial cells.20 Suppression of CXCL16/CXCR6 axis reduced LPS-induced acute lung injury.21 Bioinformatic analysis has shown that CXCL3 was a differentially expressed gene in mechanical ventilation and LPS-induced mice with acute lung injury, and might be regarded as a potential target for the treatment of acute lung injury.18 This is the first evidence demonstrating the contribution of CXCL3 to LPS-induced cytotoxicity and inflammation in BEAS-2B and HPAE cells. Moreover, knockdown of CXCL3 reduced apoptosis and inflammation in LPS-treated BEAS-2B and HPAE cell lines, thus ameliorating sepsis-associated acute lung injury.
LPS caused the reduction of viability in BEAS-2B, promoted the release of lactate dehydrogenase and induced excessive accumulation of inflammatory factors, such as TNF-α, IL-6, and TGF-β, thus contributing to the development of acute lung injury.22 LPS also induced inflammatory responses, endothelial injury, and accumulation of reactive oxygen species in HPAEC.23 Therefore, LPS-treated BEAS-2B and HPAE cell lines are widely used as cell models for sepsis-associated acute lung injury. In this study, BEAS-2B and HPAE cells were treated with LPS, which induced cytotoxicity in the cells by cell viability reduction, increase in cell apoptosis, and upregulation of TNF-α, IL-1β, and IL-18.
A previous study has shown that CXCL3 was upregulated in lung tissues of mechanical ventilation and LPS-induced mice.18 Here, LPS also induced upregulation of CXCL3 in BEAS-2B and HPAE cells. Functional assays showed that knockdown of CXCL3 decreased viability, increased apoptosis, and upregulated TNF-α, IL-1β, and IL-18 in LPS-treated BEAS-2B and HPAE cells. Therefore, knockdown of CXCL3 might exert antiapoptotic and anti-inflammatory effects against sepsis-associated acute lung injury, which needs further investigation.
MAPKs signaling has been shown to be essential for dysregulated and overly aggressive inflammatory response in the development of sepsis.24 MAPKs were abnormally activated in rats with sepsis-associated acute lung injury,25 and inhibition in the phosphorylation of p38 and JNK in lung tissues alleviated sepsis-associated acute lung injury.24 Activation of CXCLs/CXCR2 axis promoted the activation of p38/ERK signaling to regulate cell survival and migration in inflammatory diseases.16 Moreover, CXCL3 activated MAPK/ERK pathway to promote tumorigenesis of uterine cervical cancer,26 and inhibitions of ERK and JNK suppressed CXCL3-induced adipogenic differentiation.27 This study indicated that overexpression of CXCL3 enhanced the phosphorylation, while knockdown of CXCL3 reduced the phosphorylation of ERK1/2, p38, and JNK in BEAS-2B and HPAE cells.
In summary, CXCL3 promoted apoptosis and inflammation, and caused upregulations of p-ERK1/2, p-p38, and p-JNK, while knockdown of CXCL3 showed opposite results in LPS-treated BEAS-2B and HPAE cells. CXCL3 might be considered as a potential target for the treatment of sepsis--associated acute lung injury. However, the effect of CXCL3 on animal models of sepsis-associated acute lung injury should be investigated in further research.
Acknowledgments
Not applicable.
Funding
Not applicable.
Competing Interests
The authors state that there are no conflicts of interest to disclose.
Ethics Approval
Not applicable.
Contribution of Authors
Yuhui Wang designed the study and carried them out. Linyan Pan supervised the data collection and analyzed and interpreted the data. Yuhui Wang and Linyan Pan prepared the manuscript for publication and reviewed the draft of the manuscript. All authors have read and approved the manuscript.
REFERENCES
1. Chuanlong Song AA, Adilijiang Kari, Abulaiti Abuduhaer. FSTL1 aggravates sepsis-induced acute kidney injury through-regulating TLR4/MyD88/NF-κB pathway in newborn rats. Signa Vitae. 2021;17(3):167–73. 10.22514/sv.2021.071
2. Gustot T. Multiple organ failure in sepsis: Prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17(2):153–9. 10.1097/MCC.0b013e328344b446
3. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–11. 10.1016/S0140-6736(19)32989-7
4. Sadowitz B, Roy S, Gatto LA, Habashi N, Nieman G. Lung injury induced by sepsis: Lessons learned from large animal models and future directions for treatment. Expert Rev Anti Infect Ther. 2011;9(12):1169–78. 10.1586/eri.11.141
5. Rittirsch D, Flierl MA, Day DE, Nadeau BA, McGuire SR, Hoesel LM, et al. Acute lung injury induced by lipopolysaccharide is independent of complement activation. J Immunol. 2008;180(11):7664–72. 10.4049/jimmunol.180.11.7664
6. Xia Y, Cao Y, Sun Y, Hong X, Tang Y, Yu J, et al. Calycosin alleviates sepsis-induced acute lung injury via the inhibition of mitochondrial ROS-mediated inflammasome activation. Front Pharmacol. 2021;12: 690549. 10.3389/fphar.2021.690549
7. Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304(3):1093–102. 10.1124/jpet.102.044487
8. Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, et al. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol. 2009;157(7):1250–62. 10.1111/j.1476-5381.2009.00297.x
9. Opal SM, Laterre P-F, Francois B, LaRosa SP, Angus DC, Mira J-P, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA. 2013;309(11):1154–62. 10.1001/jama.2013.2194
10. Tidswell M, Tillis W, LaRosa SP, Lynn M, Wittek AE, Kao R, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. 2010;38(1):72–83. 10.1097/CCM.0b013e3181b07b78
11. Rice TW, Wheeler AP, Bernard GR, Vincent J-L, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo--controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38(8):1685–94. 10.1097/CCM.0b013e3181e7c5c9
12. Esche C, Stellato C, Beck LA. Chemokines: Key Players in Innate and Adaptive Immunity. J Invest Dermatol. 2005;125(4):615–28. 10.1111/j.0022-202X.2005.23841.x
13. Meng L, Cao H, Wan C, Jiang L. MiR-539-5p alleviates sepsis-induced acute lung injury by targeting ROCK1. Folia Histochem Cytobiol. 2019;57(4):168–78. 10.5603/FHC.a2019.0019
14. Jin L, Batra S, Douda DN, Palaniyar N, Jeyaseelan S. CXCL1 contributes to host defense in polymicrobial sepsis via modulating T cell and neutrophil functions. J Immunol. 2014;193(7):3549–58. 10.4049/jimmunol.1401138
15. Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J Clin Invest. 2007;117(3):549–56. 10.1172/JCI30562
16. Cheng Y, Ma X-l, Wei Y-q, Wei X-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev. 2019;1871(2):289–312. 10.1016/j.bbcan.2019.01.005
17. Sokulsky LA, Garcia-Netto K, Nguyen TH, Girkin JLN, Collison A, Mattes J, et al. A critical role for the CXCL3/CXCL5/CXCR2 neutrophilic chemotactic axis in the regulation of type 2 responses in a model of rhinoviral-induced asthma exacerbation. J Immunol. 2020;205(9):2468. 10.4049/jimmunol.1901350
18. Dong W-W, Feng Z, Zhang Y-Q, Ruan Z-S, Jiang L. Potential mechanism and key genes involved in mechanical ventilation and lipopolysaccharide-induced acute lung injury. Mol Med Rep. 2020;22(5):4265–77. 10.3892/mmr.2020.11507
19. Yan Z, Xiaoyu Z, Zhixin S, Di Q, Xinyu D, Jing X, et al. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci Rep. 2016;6(1):20156. 10.1038/srep20156
20. Wang X, Zhao Z, Zhu K, Bao R, Meng Y, Bian J, et al. Effects of CXCL4/CXCR3 on the lipopolysaccharide-induced injury in human umbilical vein endothelial cells. J Cell Physiol. 2019;234(12):22378–85. 10.1002/jcp.28803
21. Tu G-W, Ju M-J, Zheng Y-J, Hao G-W, Ma G-G, Hou J-Y, et al. CXCL16/CXCR6 is involved in LPS-induced acute lung injury via P38 signalling. J Cell Mol Med. 2019;23(8):5380–9. 10.1111/jcmm.14419
22. Chen L, Xu J, Deng M, Liang Y, Ma J, Zhang L, et al. Telmisartan mitigates lipopolysaccharide (LPS)-induced production of mucin 5AC (MUC5AC) through increasing suppressor of cytokine signaling 1 (SOCS1). Bioengineered. 2021;12(1):3912–23. 10.1080/21655979.2021.1943605
23. He M, Shi W, Yu M, Li X, Xu J, Zhu J, et al. Nicorandil attenuates LPS-induced acute lung Injury by pulmonary endothelial cell protection via NF-κB and MAPK pathways. Oxid Med Cell Longev. 2019;2019:4957646. 10.1155/2019/4957646
24. Fang W, Cai SX, Wang CL, Sun XX, Li K, Yan XW, et al. Modulation of mitogen-activated protein kinase attenuates sepsis-induced acute lung injury in acute respiratory distress syndrome rats. Mol Med Rep. 2017;16(6):9652–8. 10.3892/mmr.2017.7811
25. Chen J, Xue X, Cai J, Jia L, Sun B, Zhao W. Protective effect of taurine on sepsis-induced lung injury via inhibiting the p38/MAPK signaling pathway. Mol Med Rep. 2021;24(3):653. 10.3892/mmr.2021.12292
26. Qi Y-L, Li Y, Man X-X, Sui H-Y, Zhao X-L, Zhang P-X, et al. CXCL3 overexpression promotes the tumorigenic potential of uterine cervical cancer cells via the MAPK/ERK pathway. J Cell Physiol. 2020;235(5):4756–65. 10.1002/jcp.29353
27. Kusuyama J, Komorizono A, Bandow K, Ohnishi T, Matsuguchi T. CXCL3 positively regulates adipogenic differentiation. J Lipid Res. 2016;57(10):1806–20. 10.1194/jlr.M067207