
Download
ORIGINAL ARTICLE
PLCE1 alleviates lipopolysaccharide-induced acute lung injury by inhibiting PKC and NF-κB signaling pathways
Xianpeng Qiua, Jijun Chena, Jinsong Lia, Linyan Panb*
aCentral Intensive Care Unit, Zigong Fourth People’s Hospital, Zigong, Sichuan Province, China
bDepartment of Respiratory Medicine, Wenzhou Central Hospital, Wenzhou, Zhejiang Province, China
Abstract
Background: Acute lung injury (ALI) is a clinical syndrome characterized by hyperosmotic pulmonary edema and increased alveolar fluid. Phospholipase C epsilon-1 (PLCE1), identified as a member of phospholipase family, and the relationship between PLCE1 and lung injury is not clear.
Objective: To assess the possible role of Phospholipase C Epsilon 1 (PLCE1) in Acute lung injury (ALI) progression and related mechanisms.
Materials and methods: The effects of LPS and PLCE1 on cell viability and apoptosis were examined by MTT and flow cytometry. Also, the level of PLCE1 was controlled by transfection of its plasmid and shRNA. The inflammatory response in response to PLCE1 overexpression or ablation was analyzed by quantitative PCR and ELISA assay. And the involvement of PKC and NF-κB signal pathway were detected by Immunoblot.
Results: In this study, we developed a LPS-induced ALI cell model. We found PLCE1 was upregulated in LPS-induced pneumonia cells and affected cell viability. Also, knockdown of PLCE1 reduced LPS-induced apoptosis of pneumonia cells. In addition, depletion of PLCE1 suppressed LPS-induced secretion of proinflammatory cytokines in pneumonia cells. Mechanically, we found depletion of PLCE1 inhibited PKC and NF-κB signal pathway, and therefore alleviated LPS-induced ALI.
Conclusion: We therefore thought PLCE1 could serve as a promising drug for ALI.
Key words: acute lung injury, phospholipase C epsilon-1, LPS, PKC, NF-κB
*Corresponding author: Linyan Pan, Department of Respiratory Medicine, Wenzhou Central Hospital, No. 252, Baili East Road, Lucheng District, Wenzhou, Zhejiang Province 325000, China. Email address: [email protected]
Received 25 January 2022; Accepted 23 February 2022; Available online 1 May 2022
Qiu X, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Acute lung injury (ALI) is a clinical syndrome characterized by hyperosmotic pulmonary edema and increased alveolar fluid.1 ALI remains a major global public health issue for clinicians. Although multiple therapeutic strategies have been developed, mortality in ALI patients remains high.2 Therefore, it is very important to explore the pathogenesis of ALI and find a new treatment strategy. Inflammation plays an important role in the pathogenesis of ALI, and inhibition of inflammation reduces the severity of ALI injury.3 Lipopolysaccharide (LPS), the main bioactive component of Gram-negative bacteria, has been widely used to induce ALI models with pathological characteristics similar to human ALI by triggering excessive inflammatory mediators.4
Suppressed protein kinase C (PKC)/Src (sarcoma)- suppressed C kinase substrate (SSeCKS) signaling pathway is involved in microvascular endothelial barrier injury, which, therefore, increases pulmonary edema.5 PKC/SSecKs-dependent destruction of polymeric fibrous actin (F-actin) in human pulmonary microvascular endothelial cells usually leads to increased vascular permeability. Nuclear factor kappa B (NF-κB) is a key transcription factor that plays an important role in mediating cell growth, differentiation, apoptosis, and inflammation.6 IkappaB (IκB) kinase is activated by phosphorylation and degradation of NF-κB family inhibitor kinase (IKK) complex. This process results in nuclear translocation of NF-κB and subsequent transcription of NF-κB-dependent genes.7
Phospholipase C epsilon-1 (PLCE1), identified as a member of phospholipase family, catalyzes the hydrolysis of membrane phospholipids to produce two important secondary messengers: diacylglycerol and inositol 1,4, 5-triphosphate.8 They, respectively, induce protein kinase C (PKC) activation and intracellular Ca2+ mobilization, which are crucial for intracellular signal transduction.7 PLCE1 mediates different external signals and has been reported to be associated with clinical staging and survival of tumors, including hepatocellular carcinoma, colorectal cancer, bladder cancer, and gastric cancer.9 In addition to playing an important role in cancer progression, PLCE1 also contributes to inflammation by activating NF-κB pathway and stimulating expressions of various inflammatory cytokines.10 However, relationship between PLCE1 and lung injury is not clear.
In this study, we found that PLCE1 was overexpressed in LPS-induced pneumonia cells, and depletion of PLCE1 could improve the viability of pneumonia cells, reduce apoptosis and secretion of pro-inflammatory factors, and alleviate LPS-induced ALI. Studies have demonstrated that PLCE1 inhibited PKC and NF-κB signaling. We, therefore, anticipated that PLCE1 could serve as a promising target for treating ALI.
Method
Cell culture
Human lung fibroblasts (HFL1) were obtained from ATCC and incubated in serum-free Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher, USA) supplied with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin in a humidified culture hood with 5% CO2 at 37°C. For LPS stimulation, cells were treated with LPS (2-, 5-, and 10-µg/mL).
MTT assay
For assessment of cell viability, cells were plated into 96-well plates at a density of 3 × 103 cells/well and performed according to Wang et al.9 Cells were treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) for cell viability detection followed by washing with phosphate buffer solution (PBS). The absorbance was assessed at 490 nm after incubation for 4 h.
Western blotting analysis
Cell lysates were collected after addition of radioimmunoprecipitation assay (RIPA) buffer. After centrifugation, protein concentration was determined by bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology, Shanghai, China). Proteins were resolved with 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were then incubated with 5% bovine serum albumin (BSA) followed by primary antibodies targeting PLCE1 (1:1,000; Thermo Scientific, Waltham, MA, USA), PKC (1:1,000; Abcam, Cambridge, UK), SSeCKS (1:1,000; Abcam), p-p65 (1:1,000; Abcam), p65 (1:1,000; Abcam), p-IκBα (1:1,000; Abcam), IκBα (1:1,000; Abcam), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:10,000; Abcam), and b-actin (1:10,000; Abcam). Membranes were maintained in horseradish peroxidase (HRP)-conjugated secondary antibodies at a ratio of 1:1,000 for 2 h after rinsing in tris buffered saline with tween (TBST) for 15 min. The signals were detected with enhanced chemiluminescent (ECL) detection kit.
Flow cytometry (FCM)
Cells were placed into 96-well plate and treated, designed, and performed according to Umar et al.10 Then the cells were fixed and labeled with 50-μg/mL propidium iodide and Annexin V for 30 min in dark. The stained cells were washed by centrifugation and resuspended and analyzed on a BD FACS Canto II (BD Biosciences).
Real-time polymerase chain reaction (RT-PCR)
Cells were isolated and used for RNA purification with total RNA kit (Tiangen, Beijing, China) and performed as demonstrated by Wang et al.9 After extraction of RNA, it was used for reverse transcription to generate complementary DNA (cDNA) after detection of RNA concentration and purity. RT-PCR was performed with SYBR-Green Master Mix (RoPLCE1, USA) and respective primers. The used primers are listed as follows:
Interleukin 1β (IL-1β): F: TGCCACCTTTTGACAGTGATG, R: AAGGTCCACGGGAAAGACAC.
Interleukin 18 (IL-18): F: TCTCCAGCAGTCCCAACTAAGC, R: AGGCAGTACAGGACAAGG.
Tumor necrosis factor-α (TNF-α): F: ACCCTCACACT CACAAACCA, R: ATAGCAAATCGGCTGACGGT.
β-actin: F: GGAGATTACTGCCCTGGCTCCTAGC, R: GGCC GGACTCATCGTACTCCTGCTT’.
The relative expression level was calculated as 2ΔΔCt.
Enzyme-linked immunosorbent serologic assay (ELISA)
The concentrations of IL-1β, TNF-α, and IL-18 in cell lysates were measured with ELISA kit, following the protocols.
Statistical Analysis
Data were presented as mean ± SD. Statistical analysis was performed using GraphPad. P < 0.05 was considered as significant.
Results
PLCE1 is induced in LPS-stimulated HFL1 cells
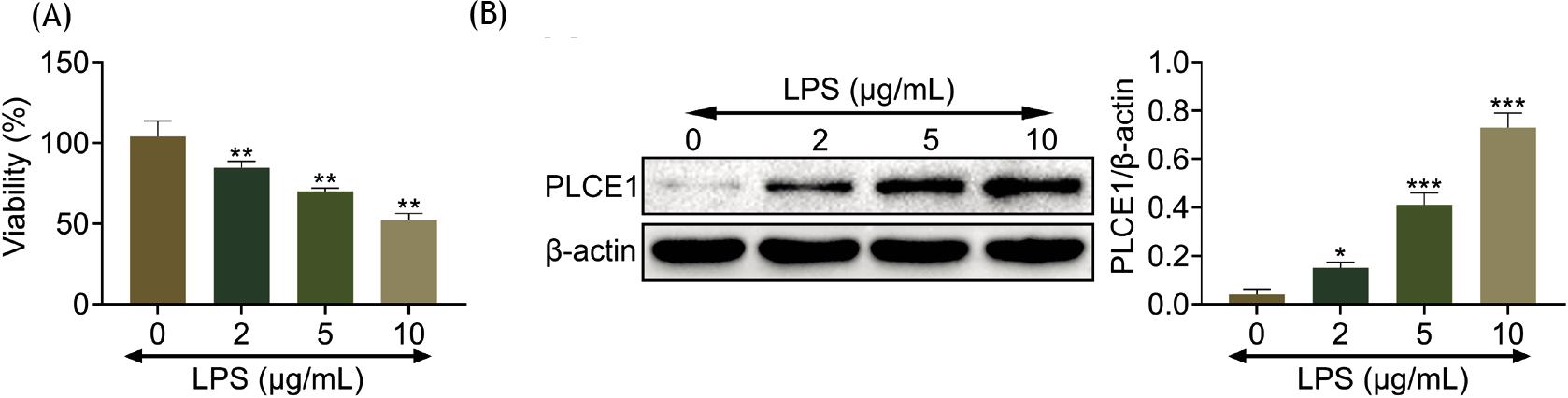
Cell viability in human fetal lung fibroblast (HFL1) cells was examined in response to increasing concentration of LPS. The cell viability was significantly reduced when stimulated with varying concentrations of LPS (Figure 1A). The expression of PLCE1 was detected when treated with LPS. Interestingly, PLCE1 was induced by LPS in HFL1 cells (Figure 1B).
Figure 1 PLCE1 is induced in LPS-stimulated HFL1 cells. (A) Cell viability in the control, LPS 2 µg/mL, LPS 5 µg/mL, and LPS 10 µg/mL group. (B) Expression level of PLCE1 in the control, LPS 2 µg/mL, LPS 5 µg/mL, and LPS 10 µg/mL group. **P < 0.01, ***P < 0.001 vs. control. LPS: lipopolysaccharide; Pc: pcDNA 3.1.
Depletion of PLCE1 improves viability of pneumonia cells, and reduces apoptosis in LPS-induced cells
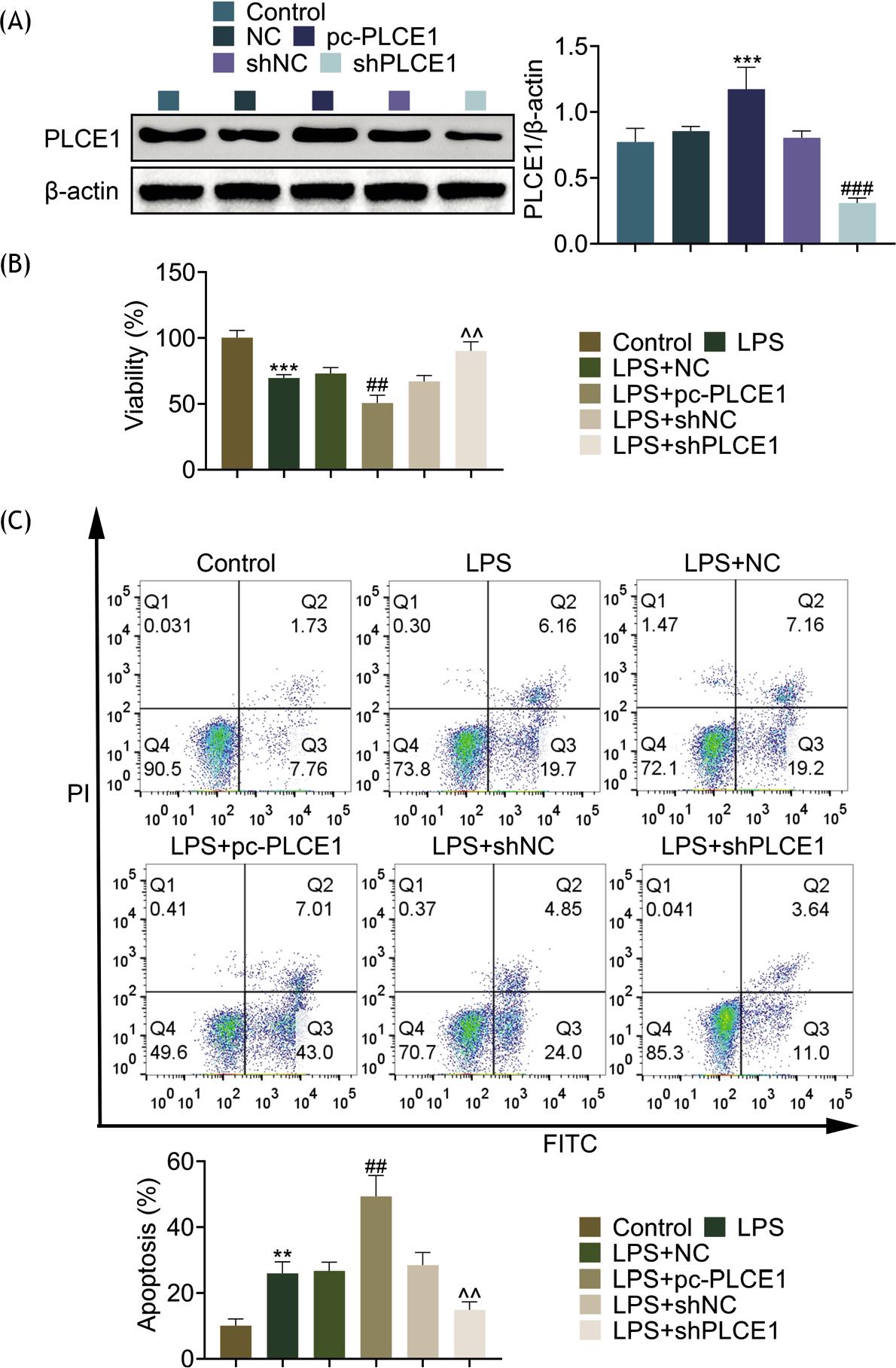
In order to further reveal the role of PLCE1 in cells, we performed PLCE1 overexpression and depletion in HFL1 cells, and the efficacy was detected by Western blotting analysis. As shown in Figure 2A, PLCE1 was induced or knocked down by transfection of PLCE1 plasmid or small hairpin RNA (shRNA). Moreover, the reduced cell viability by LPS was further diminished by overexpression of PLCE1. Conversely, the reduced viability was recovered by depletion of PLCE1 (Figure 2B). Moreover, its role in cell apoptosis was also measured. The induced cell apoptosis by LPS was aggravated by PLCE1 overexpression and relieved by PLCE1 depletion (Figure 2C). These data suggest the beneficial role of PLCE1 depletion in cell viability and apoptosis.
Figure 2 Depletion of PLCE1 could improve the viability of pneumonia cells and reduce apoptosis in LPS-induced cells. (A) Level of PLCE1 in control, NC, pc-PLCE1, shNC, and shPLCE1 cells. (B) Cell viability in control, NC, pc-PLCE1, shNC, and shPLCE1 cells. (C) Cell viability in control, NC, pc-PLCE1, shNC, and shPLCE1 cells. *P < 0.05, **P < 0.01, ***P < 0.001. LPS: lipopolysaccharide; Pc: pcDNA 3.1.
Depletion of PLCE1 could reduce secretion of pro-inflammatory factors in LPS-stimulated cells
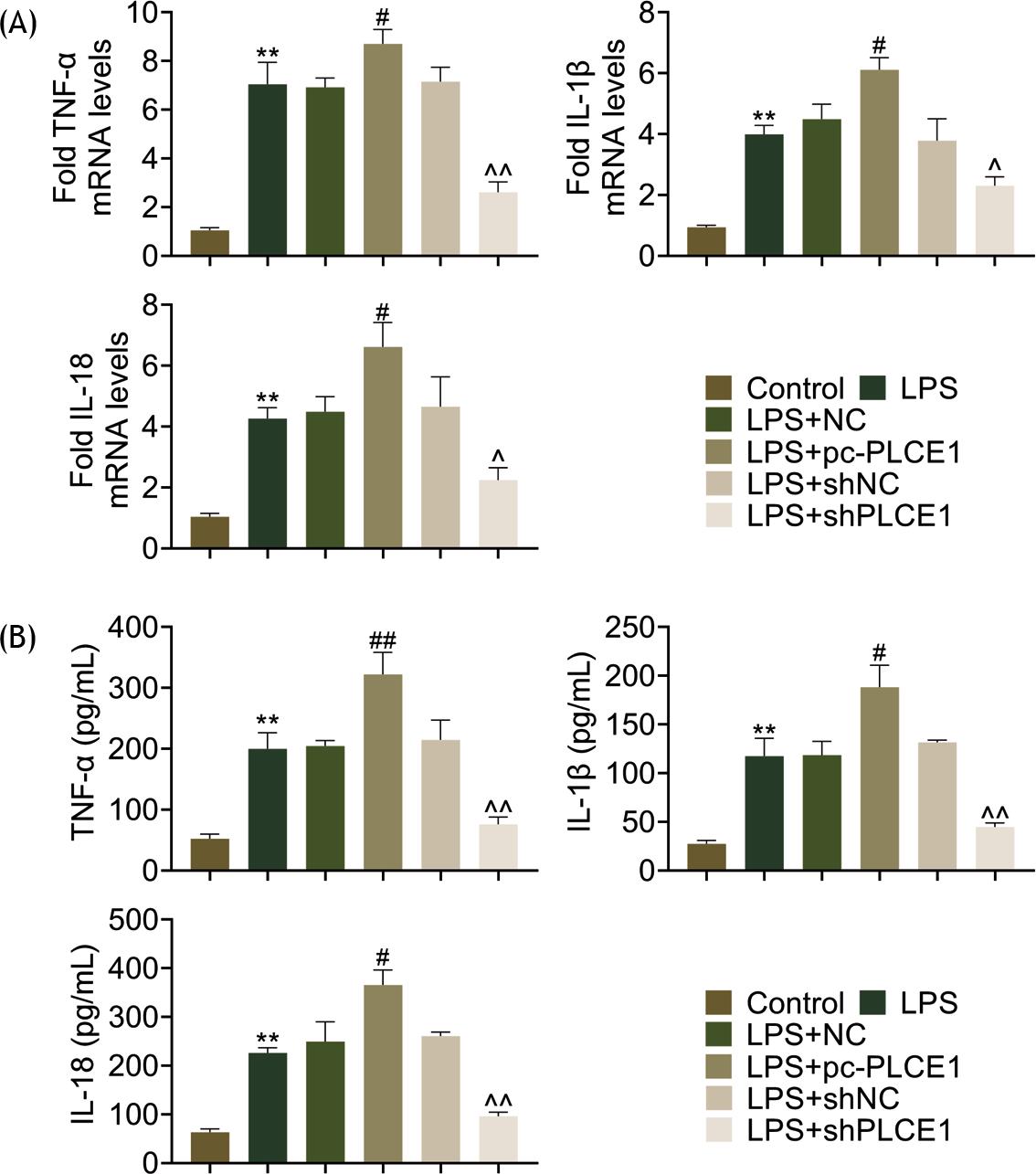
In order to explore inflammatory response in an LPS-induced cell model, levels of TNF-α, IL-1β, and IL-18 were measured in cells. The mRNA level and concentration of these cytokines were elevated by LPS, and PLCE1 overexpression further exacerbated the elevation of cytokines. PLCE1 ablation improved inflammatory response as identified by reduction of these cytokines (Figures 3A and B).
Figure 3 Depletion of PLCE1 could reduce secretion of pro-inflammatory factors in LPS-stimulated cells. (A) mRNA and (B) proteinlevels of TNF-a, IL-1b, and IL-18 in control, NC, pc-PLCE1, shNC, and shPLCE1 cells. *P < 0.05, **P < 0.01, ***P < 0.001. LPS: lipopolysaccharide; pc: pcDNA 3.1.
PLCE1 inhibits PKC and NF-κB signaling in pneumonia cells
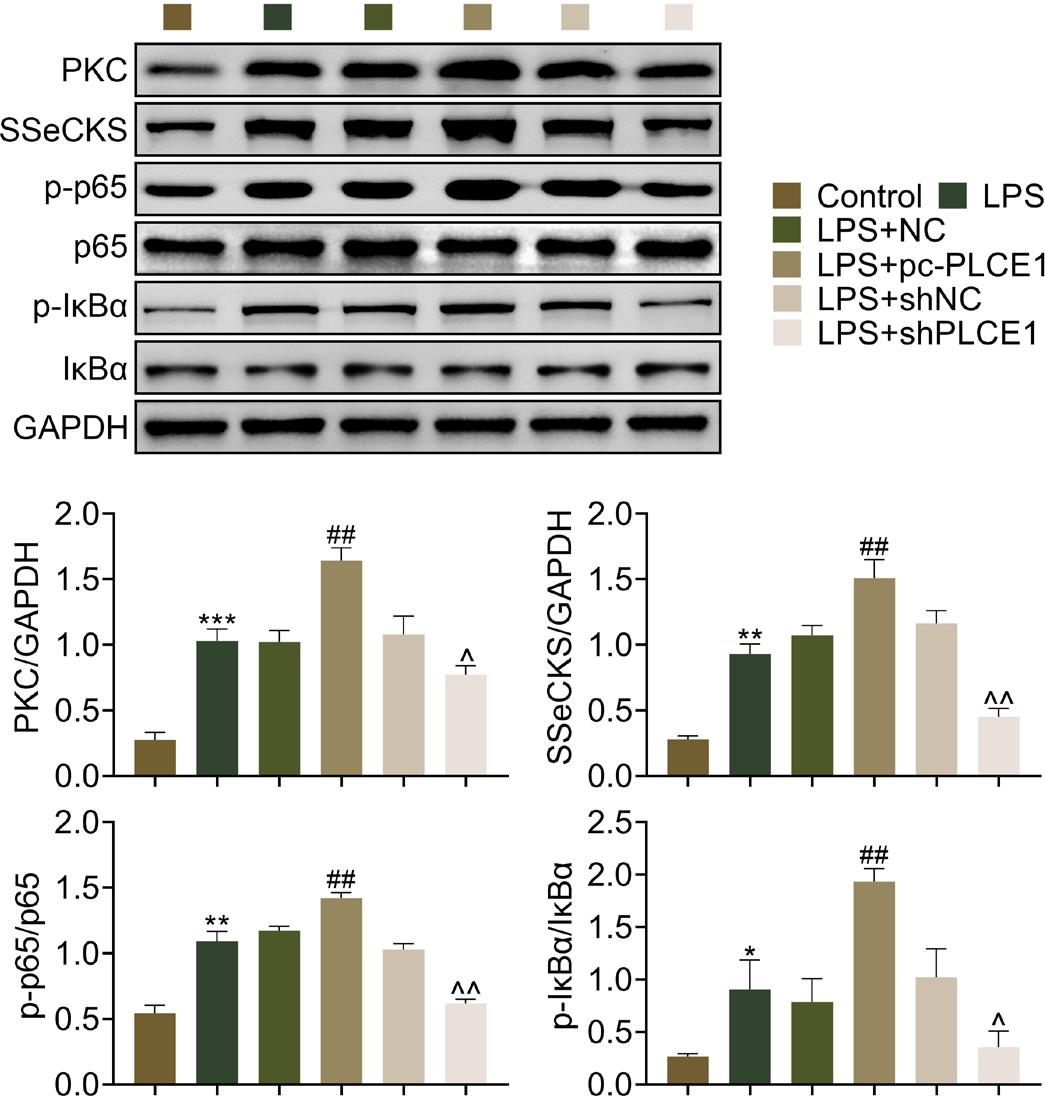
In order to delineate the potential mechanism of PLCE1-mediated cell proliferation and apoptosis, the inhibition of PKC and NF-κB signaling was analyzed in each group. The expression levels of PKC, SSeCKS, p-p65, and p-IκBα were enhanced in LPS-induced cells (Figure 4). Overexpression of PLCE1 further elevated activation of these proteins, and PLCE1 depletion led to repression of PKC, SSeCKS, p-p65, and p-IκBα levels (Figure 4). These data suggested that PLCE1 inhibited PKC and NF-κB signal pathway, and therefore alleviated LPS-induced ALI.
Figure 4 PLCE1 inhibits PKC and NF- κB signaling in pneumonia cells. Levels of PKC, SSeCKS, p- p65, and p- IκBα in control, NC, pc- PLCE1, shNC, and shPLCE1 cells. *P < 0.05, **P < 0.01, ***P < 0.001. LPS: lipopolysaccharide; pc: pcDNA 3.1.
Discussion
Acute lung injury is injury of the alveolar epithelial and endothelial cells caused by various direct and indirect factors.11 The treatment of ALI mainly includes respiratory support therapy and drug therapy. Although some progress has been observed in treatment regimen, its mortality rate is still high. In order to improve the survival rate, it is necessary to further study the pathogenesis of this disease and identify key proteins.12 Inflammation plays an important role in the pathogenesis of ALI. Inhibition of inflammation leads to reduction of the severity of ALI injury.13 In this study, we developed an LPS-induced ALI cell model, and found that PLCE1 played a crucial role in the progression of ALI. We noticed that PLCE1 could alleviate LPS-induced ALI.
Through a series of in vitro assays, including Immunoblot, MTT, FCM, qPCR, and ELISA, we found that PLCE1 was overexpressed in LPS-induced pneumonia cells. Knock down of PLCE1 could improve the viability of pneumonia cells and reduce apoptosis and secretion of proinflammatory factors to alleviate LPS-induced ALI. In fact, the role of PLCE1 in different cellular processes has been revealed. Its expression was correlated with several types of cancers, such as esophageal and liver cancers.14,15 PLCE1 could also mediate the migration, proliferation as well as differentiation of podocytes.16 In addition, PLCE1 affected progression of esophageal squamous cell carcinoma and could promote myocardial ischemia-reperfusion injury (MI-RI) in H9C2 cells and mice by stimulating inflammation.17,19 Similarly, we also revealed that PLCE1 mediated inflammation in LPS-induced ALI model.
In this study, we found that PLCE1 inhibited PKC and NF-κB pathway in LPS-induced pneumonia cells. In fact, the role of PKC and NF-κB pathway in ALI has been widely revealed. For example, depletion of PKC attenuated oleic acid-induced acute lung injury through reduction of inflammation and oxidative stress. In addition, the role of PKC and NF-κB pathways in the regulation of inflammation has also been widely accepted. Sulfur could suppress high glucose-induced inflammation via mediating NF-κB pathway in monocytes.18 PKC could also mediate LPS-Induced IL-1β expression and affect the pro-inflammatory effect of androgen receptor (AR) in mouse microglia.19 In addition, another study indicated that Cathepsin C contributed to the microglia M1 polarization and aggravated neuroinflammation by the activation of Ca-dependent PKC/NF-κB pathway.20 These studies confirmed that PKC and NF-κB pathway could serve as promising targets for inflammation-related diseases.
Conclusion
Here, we developed an ALI cell model by using LPS. Notably, LPS-induced pneumonia cells are widely used in the study of ALI. We revealed the effects of PLCE1 on LPS-induced pneumonia cells, and revealed the regulatory mechanism.
In summary, we found the overexpression of PLCE1 in LPS-induced pneumonia cells. Our data confirmed that PLCE1 could promote the viability of LPS-induced pneumonia cells. PLCE1 could also reduce the apoptosis and secretion of pro-inflammatory factors, and alleviate LPS-induced ALI. Mechanically, PLCE1 inhibited PKC and NF-κB signaling pathways, and therefore mediated ALI progression. We, therefore, anticipated that PLCE1 could serve as a promising target of ALI.
Competing interests
The authors stated that there were no conflicts of interest to disclose.
Contribution of authors
Xianpeng Qiu and Jijun Chen designed and carried the experiments. Jinsong Li analyzed and interpreted the data. Linyan Pan prepared the manuscript with contributions from all coauthors.
REFERENCES
1. Yang J, Do-Umehara HC, Zhang Q, Wang H, Hou C, Dong H, et al. miR-221-5p-mediated downregulation of JNK2 aggravates acute lung injury. Front Immunol. 2021;12:700933. 10.3389/fimmu.2021.700933
2. Nelin LD, Jin Y, Chen B, Liu Y, Rogers LK, Reese J. Cyclooxygenase-2 deficiency attenuates lipopolysaccharide-induced inflammation, apoptosis and acute lung injury in adult mice. Am J Physiol Regul Integr Comp Physiol. 2022, Feb 1;322(2):R126-35. 10.1152/ajpregu.00140.2021
3. Martin AK. Predicting the price of the pump: Examining the incidence and predictive factors of acute kidney injury following off-pump lung transplantation. J Cardiothorac Vasc Anesth. 2022;36(1):100–2. 10.1053/j.jvca.2021.10.031
4. Jin X, Gao X, Lan M, Li CN, Sun JM, Zhang H. Study the mechanism of peimisine derivatives on NF-kappa-B inflammation pathway in mice with acute lung injury induced by lipopolysaccharide. Chem Biol Drug Design. 2021, Dec 28. Online ahead of print. 10.1111/cbdd.14013
5. Li Y, Luan C. PLCE1 promotes the invasion and migration of esophageal cancer cells by up-regulating the PKCalpha/NF-kappaB pathway. Yonsei Med J. 2018;59(10):1159–65. 10.3349/ymj.2018.59.10.1159
6. Chen Y, Wang D, Peng H, Chen X, Han X, Yu J, et al. Epigenetically upregulated oncoprotein PLCE1 drives esophageal carcinoma angiogenesis and proliferation via activating the PI-PLCepsilon-NF-kappaB signaling pathway and VEGF-C/Bcl-2 expression. Mol Cancer. 2019;18(1):1. 10.1186/s12943-018-0930-x
7. Bye H, Prescott NJ, Lewis CM, Matejcic M, Moodley L, Robertson B, et al. Distinct genetic association at the PLCE1 locus with oesophageal squamous cell carcinoma in the South African population. Carcinogenesis. 2012;33(11):2155–61. 10.1093/carcin/bgs262
8. Sharma KL, Umar M, Pandey M, Misra S, Kumar A, Kumar V, et al. Association of potentially functional genetic variants of PLCE1 with gallbladder cancer susceptibility in north Indian population. J Gastrointest Cancer. 2013;44(4):436–43. 10.1007/s12029-013-9537-z
9. Wang Q, Chen P, Chen D, Liu F, Pan W. Association between phospholipase C epsilon gene (PLCE1) polymorphism and colorectal cancer risk in a Chinese population. J Int Med Res. 2014;42(2):270–81. 10.1177/0300060513492484 10.1177/0300060514559792
10. Umar M, Upadhyay R, Kumar S, Ghoshal UC, Mittal B. Role of novel and GWAS originated PLCE1 genetic variants in susceptibility and prognosis of esophageal cancer patients in northern Indian population. Tumour Biol. 2014;35(11):11667–76. 10.1007/s13277-014-2458-z
11. Herrlich A. Interorgan crosstalk mechanisms in disease: The case of acute kidney injury-induced remote lung injury. FEBS Lett. 2021. Online ahead of print. doi: 10.1002/1873-3468.14262
12. Chen W, Chen H, Yang ZT, Mao EQ, Chen Y, Chen EZ. Free fatty acids-induced neutrophil extracellular traps lead to dendritic cells activation and T cell differentiation in acute lung injury. Aging. 2021;13(24):26148–60. 10.18632/aging.203802
13. Song J, Chen D, Pan Y, Shi X, Liu Q, Lu X, et al. Discovery of a novel MyD88 inhibitor M20 and its protection against sepsis-mediated acute lung injury. Front Pharmacol. 2021;12:775117. 10.3389/fphar.2021.775117
14. Sun H, Wu X, Wu F, Li Y, Yu Z, Chen X, et al. Associations of genetic variants in the PSCA, MUC1 and PLCE1 genes with stomach cancer susceptibility in a Chinese population. PloS One. 2015;10(2):e0117576. 10.1371/journal.pone.0117576
15. Malik MA, Srivastava P, Zargar SA, Mittal B. Phospholipase C epsilon 1 (PLCE1) haplotypes are associated with increased risk of gastric cancer in Kashmir valley. Saudi J Gastroenterol. 2014;20(6):371–7. 10.4103/1319-3767.145330
16. Li Y, An J, Huang S, Liao H, Weng Y, Cai S, et al. PLCE1 suppresses p53 expression in esophageal cancer cells. Cancer Investig. 2014;32(6):236–40. 10.3109/07357907.2014.905588
17. Qu Y, Zhang S, Cui L, Wang K, Song C, Wang P, et al. Two novel polymorphisms in PLCE1 are associated with the susceptibility to esophageal squamous cell carcinoma in Chinese population. Dis Esophagus. 2017;30(1):1–7. 10.1111/dote.12463
18. Shekarriz R, Faghani S, Tafazoli A, Hashemi-Soteh MB. The correlation between phospholipase C epsilon (PLCE1) gene polymorphisms and risk of gastric adenocarcinoma in Iranian population. Int J Hematol Oncol Stem Cell Res. 2019;13(3): 108–15. 10.18502/ijhoscr.v13i3.1268
19. Li W, Li Y, Chu Y, Wu W, Yu Q, Zhu X, et al. PLCE1 promotes myocardial ischemia-reperfusion injury in H/R H9c2 cells and I/R rats by promoting inflammation. Biosci Rep. 2019;39(7):BSR20181613. 10.1042/BSR20181613
20. Hu X, Jia J, Yang Z, Chen S, Xue J, Duan S, et al. PLCE1 polymorphisms are associated with gastric cancer risk: The changes in protein spatial structure may play a potential role. Front Genet. 2021;12:714915. 10.3389/fgene.2021.714915