
Download
ORIGINAL ARTICLE
Demographic and clinical characterization of pediatric group patients with inborn errors of the immune system in a Colombian tertiary hospital
Manuela Olayaa, b*, Daniela Clevesb, Tania Guzmánb, c, Laura Torres-Canchalac, Harry Pachajoab, d, Diego Medina-Valenciae, f, Jaime Patiñob, g, José David Gómezb, Paola Pérezb, g
aPediatric Allegology Service, Fundación Valle del Lili, Cali, Colombia
bFacultad de Ciencias de la Salud, Universidad Icesi, Cali, Colombia
cCentro de Investigaciones Clínicas, Fundación Valle del Lili, Cali, Colombia
dCentro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER), Universidad Icesi, Cali, Colombia
eClinical Genetic Service, Fundación Valle del Lili. Cali, Colombia
fBone Marrow Transplant Unit, Fundación Valle del Lili, Cali, Colombia
gPediatric Infectious Diseases Service, Fundación Valle del Lili, Cali, Colombia
Abstract
Introduction: In recent decades, there has been a growing increase in the diagnosis of patients with inborn errors of the immune system, formerly known as primary immunodeficiency disorders (PIDs). Timely diagnosis remains a challenge due to low clinical suspicion and poor education on the subject. It is estimated that between 70% and 90% of these pathologies remain underdiagnosed in our environment.
Objective: The objective of this study is to characterize the demographic and clinical presentation of pediatric group patients with inborn errors of the immune system in a Colombian tertiary hospital.
Methods: Retrospective descriptive study of 306 patients with a diagnosis of innate errors of the immune system who consulted the PID clinic between 2011 and 2018 in a high-complexity institution in Cali, Colombia.
Results: Three-hundred and six patients were included. The median age was 4 years (IQR 2.3–7.7 years), and 59.5% of the patients were male. According to the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency classification for inborn errors of the immune system, the most common group was antibody deficiency in 74.8% (n˂229), especially in the age group between 1 and 5 years. The least frequent in our population was complement deficiency. Of the warning signs stipulated for these pathologies, the most frequent were the (1) need for intravenous antibiotics (32%), (2) difficulty growing (15.7%), (3) four or more episodes of ear infection (10.8%), and (4) abscesses in organs or cutaneous abscesses (12.7%). No patient reported two or more episodes of pneumonia or sinusitis, and only 5.8% of the patients received a bone marrow transplant.
Conclusions: Innate errors of the immune system require an early diagnosis with follow-up from an early age to ensure adequate management and follow-up in order to reduce morbidity and mortality. It is imperative to sensitize the medical population about the existence of these pathologies so that early intervention can be carried out, which improves the quality of life of patients and their families.
Key words: Primary immunodeficiency disorders, innate errors of the immune system, antibody deficiency, Tertiary hospital, Colombia
*Corresponding author: Manuela Olaya, Pediatric Allegology Service and Facultad de Ciencias de la Salud, Fundación Valle del Lili, Carrera. 98 # 18-49, Cali, Colombia. Email address: [email protected]
Received 5 November 2020; Accepted 2 December 2021; Available online 1 July 2022
Copyright: Olaya M, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Inborn errors of immunity, previously known as primary immunodeficiency disorders (PIDs), are a heterogeneous group of genetic disorders caused by defects in the development or function of the immune system. Most of them present at an early age as recurrent or severe infections, malignancy, or dysregulation in the immune response; autoinflammation; autoimmunity; or allergic disease.1,2
In 1970, the World Health Organization created a committee to classify these pathologies.3 Subsequently, in 2015, the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency (IUIS) modified the classification, given the recent advances of the last 40 years, with the aim to increase awareness, facilitate recognition, standardize nomenclature, standardize the approach, and promote optimal and early treatment for these patients.4 In this classification, nine groups based on the immunophenotype were established to homogenize diagnoses.4,5 Subsequently, this classification was updated in 2019 to include more pathologies and defects.6 Currently, more than 400 genetic defects associated with the development of innate errors of the immune system have been described.6–8
The Jeffrey Modell Foundation (JMF) published an expert consensus of the 10 warning signs for PIDs that help identify patients in primary care who should be suspected of these pathologies. If the patient presents two or more of the following signs, this should suggest the presence of innate errors of the immune system: (1) four or more ear infections within 1 year, (2) two or more serious sinus infections within 1 year, (3) two or more episodes of ineffective response to antibiotic management, (4) two or more episodes of pneumonia within 1 year, (5) failure to grow normally, (6) recurrent skin abscesses or abscesses in organs, (7) persistent canker sores in the mouth or fungal infection on the skin, (8) need for intravenous antibiotics to eradicate infections, (9) two or more systemic or deep infections, and (10) the presence of a family history of PIDs.9
Different countries in Europe and Asia report prevalence of innate errors of the immune system between 1:8500 and 1:100,000, which is higher in populations where inbreeding is common.10–14 In the United States, prevalence between 29.1 and 50.5 patients has been reported for each 100,000 inhabitants.11 In Latin America, a regional database was created in 2009 by the Latin American Society for Primary Immunodeficiencies (Society for Immunodeficiencies Database or LASID). This database reports a prevalence of 1 case per 20,000 inhabitants in Argentina and up to 1 case per 146,000 inhabitants in Peru.15 In Colombia, the estimated prevalence in 2020 was 2.07 cases per 50.000 inhabitants.16
Timely diagnosis continues to be a challenge due to low clinical suspicion and poor education on the subject. It is estimated that between 70% and 90% of innate errors of the immune system remain underdiagnosed in our environment.15 The objective of this study is to characterize the demographic and clinical presentation of pediatric group patients with inborn errors of the immune system in a Colombian tertiary hospital.
Materials and Methods
A retrospective, descriptive study was carried out. Patients under 18 years of age with a diagnosis of inborn errors of immunity treated between 2011 and 2018 at a highly complex institution in southwestern Colombia were included.
The information was obtained from institutional statistics of patients under 18 years of age who had a diagnosis with any of the ICD-10: D80-D89, in addition to the records of the pediatric infectious disease and pediatric immunology services. The diagnosis of inborn errors of the immune system is made based on clinical and paraclinical criteria, which are based on the European Society for Immunodeficiencies classification.17 In addition, the IUIS classification of 2015 was used for immunotyping by subgroups.4 We found 306 patients who met the inclusion criteria.
Subsequently, a review of the medical records was carried out by two medical students trained in the definitions of the protocol, and a database was completed in the institutional platform for clinical studies, BD-Clinic. Sociodemographic, clinical, paraclinical, and management variables were included.
Statistical analysis
A univariate analysis was carried out to determine the behavior of the numerical variables, determining the normality of the variables through a Shapiro–Wilk test, and those with a normal distribution were presented with measures of central tendency as means and standard deviation. Those measures that were not normally distributed were presented with median and interquartile ranges. Categorical variables are presented as proportions and percentages. To assess data quality, inconsistent or extreme data were checked against the source document. The analysis was performed using the statistical package Stata 14.0 (StataCorp, College Station, TX, USA).
Target organ involvement was analyzed, including lung involvement (presence of bronchiectasis, atelectasis, and/or interstitial lung disease), liver involvement (evidence of fibrosis and/or elevation of transaminases), and skin involvement (presence of pigmentary alterations, hair alterations and nails, warts, seborrheic dermatitis, molluscum contagiosum, chronic urticaria, prurigo, skin infections, and/or atopic dermatitis). The outcomes including bone marrow transplantation (BMT), intravenous and subcutaneous immunoglobulin replacement, and antimicrobial prophylaxis (trimethoprim sulfamethoxazole [TMS]––fluconazole–acyclovir) were characterized.
Results
Three-hundred and six pediatric patients with a diagnosis of innate error of the immune system who visited our institution between 2011 and 2018 were included. . The median age was 4 years (IQR 2.3–7.7 years), 59.5% of the patients were male, and 72.7% were older than 5 years (Table 1). According to the IUIS classification, the most common group was predominantly antibody deficiency in 75.2% (n˂230), especially in the age group between 1 and 5 years (Table 1). The second most frequent group was combined immunodeficiency (CID) associated with well-defined syndromes in 22 patients (7.2%). The least frequent in our population was complement deficiency.
Table 1 Different innate errors of the immune system according to the IUIS 2015 classification in patients consulting the PID clinic in FVL between 2011 and 2018 according to age group % (n).
| Characteristics | ˂1 year | 1–5 years | ˃5 years | Total N=306 |
|---|---|---|---|---|
| Gender, n (%) | ||||
| Female | 18 (60) | 79 (44.6) | 27 (27.3) | 124 (40.5) |
| Male | 12 (40) | 98 (55.4) | 72 (72.7) | 182 (59.5) |
| IUIS classification, n (%) | ||||
| Predominantly antibody deficiencies | 16 (53.3) | 141 (79.7) | 73 (73.7) | 230 (75.2) |
| CID with associated or syndromic features | 3 (10) | 7 (4) | 12 (12.1) | 22 (7.2) |
| Immunodeficiencies affecting cellular and humoral immunity | 5 (16.7) | 11 (6.2) | 5 (5.1) | 21 (6.9) |
| Congenital defects of phagocyte number, function, or both | 5 (16.7) | 10 (5.6) | 1 (1) | 16 (5.2) |
| Autoinflammatory disorders, | 0 (0) | 3 (1.7) | 4 (4) | 7 (2.3) |
| Disease of immune dysregulation | 0 (0) | 3 (1.7) | 2 (2) | 5 (1.6) |
| Defects in intrinsic and innate immunity | 1 (3.3) | 2 (1.1) | 0 (0) | 3 (1) |
| Complement deficiencies | 0 (0) | 0 (0) | 2 (2) | 2 (0.7) |
According to the IUIS classification, the majority of our patients had predominant antibody deficiencies, of which 79.1% had nonspecific hypogammaglobulinemia. The second most frequent classification was CID with associated or syndromic features within which 40.9% presented hyper IgE syndrome (Table 2).
Table 2 Immunodeficiency diagnosis according to IUIS classification.
| N=306 | |
|---|---|
| Predominantly antibody deficiencies | n=230 (%) |
| Nonspecific hypogammaglobulinemia | 182 (79.1) |
| Selective IgA deficiency | 26 (11.3) |
| Specific antibody deficiency | 5 (2.2) |
| Common variable immunodeficiency disorders | 13 (5.7) |
| Agammaglobulinemia | 4 (1.7) |
| CID with associated or syndromic features | n=22 |
| Hyper IgE syndrome (HIES) | 9 (40.9) |
| Ataxia telangiectasia | 3 (13.6) |
| Wiskott–Aldrich syndrome | 2 (9.1) |
| DiGeorge syndrome | 1 (4.5) |
| Congenital dyskeratosis | 2 (9.1) |
| Hyper IgM syndrome | 5 (22.7) |
| Immunodeficiencies affecting cellular and humoral immunity | n=21 |
| Lymphopenia CD4 | 9 (42.9) |
| Lymphopenia CD8 | 2 (9.5) |
| Lymphopenia NK | 2 (9.5) |
| No severe combined immunodeficiency | 4 (19) |
| Severe combined immunodeficiency | 4 (19) |
| Congenital defects of phagocyte number, function, or both | n=16 |
| Congenital neutropenia | 14 (87.5) |
| Chronic granulomatous disease | 1 (6.3) |
| Papillon–Lefèvre syndrome | 1 (6.3) |
| Autoinflammatory disorders | n=7 |
| Haploinsufficiency A20 | 1 (14.3) |
| Familial Mediterranean fever | 6 (85.7) |
| Disease of immune dysregulation | n=5 |
| Hemophagocytic lymphohistiocytosis syndromes | 2 (40) |
| Autoimmune lymphoproliferative syndrome (alps) | 2 (40) |
| Early onset inflammatory disease | 1 (20) |
| Defects in intrinsic and innate immunity | n=3 |
| Chronic mucocutaneous candidiasis | 1 (33.3) |
| Systemic papillomatosis | 1 (33.3) |
| Herpetic encephalitis | 1 (33.3) |
| Complement deficiencies | n=2 |
| C4 Complement deficiency | 2 (100) |
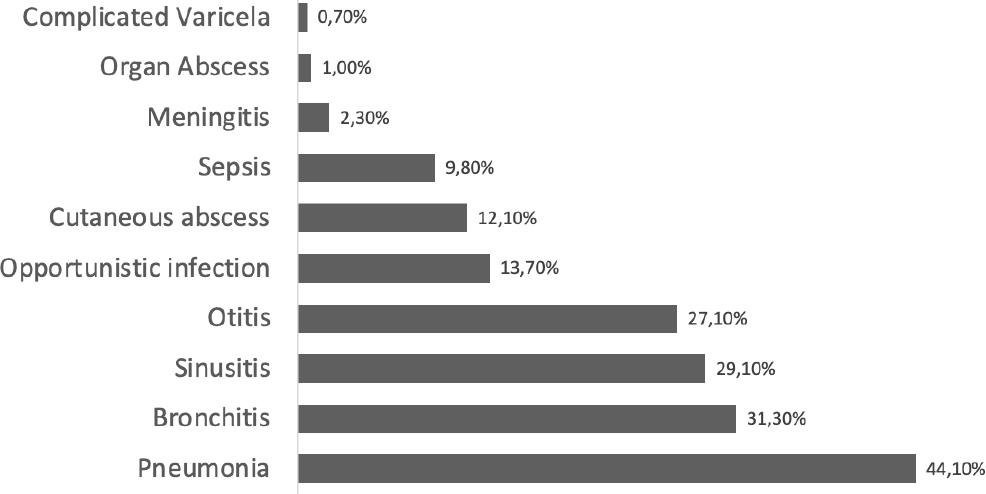
Pneumonia was the most frequent infectious disease prior to diagnosis, evidenced in 135 patients (44.1%), followed by recurrent episodes of bronchitis (31.3%). No patient reported two or more episodes of pneumonia. It is also worth mentioning that 27.1% of the patients had otitis prior to diagnosis [median of episodes prior to diagnosis of 3 IQR (1-4)]. (Figure 1). We found that 66.3% (203) of all patients admitted with PID diagnosis had allergic diseases.
Figure 1 Infectious events prior to diagnosis of inborn errors of the immune system in pediatric patients consulting the PID clinic in FVL between 2011 and 2018.
Of the warning signs stipulated by the JMF, the most frequent were (1) the need for intravenous antibiotics (32%), (2) two or more pneumonia episodes (22.2%), (3) two or more sinusitis episodes (20.3%), and (4) growth difficulty (15.7%) (Table 3). The difference in the order of the red flags between the two most frequent groups according to the IUS 2015 classification varies in those patients with CID with associated or syndromic features, and the third place corresponds to those patients with four or more episodes of otitis. Prior to diagnosis, 60.1% (n=84) of patients had alarm signs.
Table 3 Different red flags according to JMF in pediatric patients with PID according to the IUIS 2015 classification during the first consultation for immunology.
| Characteristics | Predominantly antibody deficiencies n=226 | CID with associated or syndromic features, n=22 | Immunodeficiencies affecting cellular and humoral immunity, n=21 | Congenital defects of phagocyte number, function, or both, n=16 | Autoinflammatory disorders, n=7 | Disease of immune dysregulation, n=5 | Defects in intrinsic and innate immunity, n=3 | Complement deficiencies, n=2 | Total, N=306 |
|---|---|---|---|---|---|---|---|---|---|
| four or more media otitis episodes | 22 (9.7) | 7 (31.8) | 1 (4.8) | 0 (0) | 1 (14.3) | 1 (20) | 0 (0) | 1 (50) | 33 (10.8) |
| two or more sinusitis episodes | 54 (23.9) | 5 (22.7) | 2 (9.5) | 0 (0) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 62 (20.3) |
| two or more pneumonia episodes | 46 (20.4) | 8 (36.4) | 11 (52.4) | 2 (12.5) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 68 (22.2) |
| Growth difficulty | 33 (14.6) | 4 (18.2) | 8 (38.1) | 1 (6.3) | 1 (14.3) | 1 (20) | 0 (0) | 0 (0) | 48 (15.7) |
| Internal organ abscess | 2 (0.9) | 0 (0) | 0 (0) | 1 (6.3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (1) |
| Intravenous antibiotics need | 66 (29.2) | 13 (59.1) | 10 (47.6) | 4 (25) | 4 (57.1) | 0 (0) | 0 (0) | 1 (50) | 98 (32) |
| Deep infections including septicemia | 11 (4.9) | 5 (22.7) | 6 (28.6) | 0 (0) | 1 (14.3) | 2 (40) | 0 (0) | 0 (0) | 25 (8.2) |
| Familiar history of PID | 3 (1.3) | 0 (0) | 0 (0) | 1 (6.3) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 5 (1.6) |
| Fungal skin infections | 6 (2.7) | 2 (9.1) | 5 (23.8) | 2 (12.5) | 0 (0) | 0 (0) | 1 (33.3) | 0 (0) | 16 (5.2) |
Among the clinical outcomes, cutaneous and pulmonary involvement was the most frequent, with 15.4% and 27.1%, respectively.
Of our patients, 28.4% (n=87) received human immunoglobulin either intravenously (IVIG) in 24.5% (n=75) or subcutaneously (IGSC) in 3.9% (n=12), 6.9% (n=21) received antifungal prophylaxis with fluconazole, 3.6% (n=11) received antiviral prophylaxis with acyclovir, and 10.5% (n=32) received antibacterial prophylaxis with TMS. Eighteen patients (5.8%) were taken to have had BMT.
Discussion
Inborn errors of the immune system are a group of disorders characterized by poor or absent function in one or more components of the immune system that predisposes affected individuals to greater frequency and severity of infections, autoimmunity, and aberrant inflammation and malignancy. The clinical presentation is highly variable, but most present as recurrent infections, which are common in childhood and therefore may go unnoticed in the primary care setting.1,2,18 We found that the most frequent group according to the IUIS 2015 classification was predominantly antibody deficiency, which is consistent with the international literature,1,9 followed by CID with associated or syndromic features and then immunodeficiencies affecting cellular and humoral immunity. LASID announced that it had a registry of 8383 patients with innate errors of the immune system by August 2020, of which 53.2% had a predominantly antibody deficiency.19 On the other hand, the groups of Pedraza et al. and Riaño-Cardozo et al. found a prevalence between 56% and 74.4% of predominantly antibody deficiency in different cities of Colombia.20,21
Inborn errors of immunity are more frequent in males due to the inheritance pattern linked to the X chromosome.22 The Jeffrey Modell Centers Network, have the registry of 44,582 patients with PID; in 2018, 58% of the patients were men.9 At the national level, a study carried out by the IDP group at the University of Antioquia found 891 patients with innate errors of the immune system, where 59.5% were male.21 This is consistent with our findings where 59.5% of our patients were male.
Regarding the warning signs for suspecting innate errors of the immune system formulated by the JMF, the most frequent signs found in our patients were the need to receive intravenous antibiotic management and two or more pneumonia episodes.
We found that 44% of our patients had presented at least one episode of pneumonia before being diagnosed with inborn errors of immunity. Something similar was found by Felgentreff et al. where the most frequent disease presented prior to the diagnosis was pneumonia, some of them of viral etiology and with severe manifestations.23 We consider that severe pneumonia in our population should be regarded as an alarm sign and should be studied since almost half of our patients presented this pathology. Knerr and Grimbacher’s findings were similar to ours.24 They found that the most common pathologies presented prior to diagnosis were pneumonia (54.9%), followed by diarrhea in 40.4%, sinusitis in 37.1%, and otitis media in 35.4%.24 The group of F. Fernández also had findings related specifically to the predominant antibody deficiency, where the most common form of presentation of the disease was recurrent infections, mainly pneumonia in 91.7% of the patients, three of whom also had a history of recurrent otitis (more than 4 per year).25
The number of infectious episodes, otitis media, sinusitis, pneumonia, and bronchitis, prior to the diagnosis of these pathologies found in this study was mostly from the respiratory tract.. The frequency of these infections and their severity were in accordance with the JMF description for suspected innate errors of immunity.9
Skin was the most frequent organ involved, with cutaneous manifestations affecting 27% of our patients, followed by lung involvement. Chronic complications of the respiratory tract are relevant: mortality from lower respiratory tract infections in these patients remains significant at 29%–44% while deaths caused by respiratory failure resulting from chronic lung disease represent 30%–36%.26
Among the patients included in the study, 22 were found to have undergone a genetic study. The remaining 286 patients did not undergo a genetic study due to administrative problems with their health entities.
Of our patients, 24.5% received IVIG and 3.9% received IGSC. A survey carried out by the JMF with different specialists in the network reported that 74.4% of the patients with diagnoses of inborn errors of the immune system received replacement of immunoglobulins and the most frequent form was the intravenous presentation.27 We consider that our data are much below these numbers because of our age group, the underdiagnosis of these entities in our environment, and administrative delays in our health system.
About 5.8% of our patients were taken to have had BMT, which is considerably lower than that reported in the literature, but this may be due to the fact that we do not have neonatal screening available, the delay in diagnosis, and, above all, the age groups of diagnosis of our patients.
Limitations
Our study has several limitations, as it is a retrospective study, there are absent data in some of the patients included, and as it may also imply that management protocols have changed over the years with the availability of new evidence. Additionally, not all of the patients had a strict follow-up in our PID clinic due to administrative problems related to the Colombian health system that delay timely allocation of appointments with specialists.
Conclusions
Inborn errors of immunity are a group of congenital diseases that affect the immune response and lead to recurrent infections with lung, liver, and skin deterioration, which is why monitoring from an early age with adequate management and monitoring is necessary to reduce long-term morbidity and mortality. It is imperative to sensitize the medical population so that they suspect these diseases and achieve a timely and appropriate intervention that improves the quality of life of patients and their families, as well as survival.
REFERENCES
1. Özdemir Ö. Current approach to primary immunodeficiency diseases. South Clin Ist Euras. 2019;30: 83–90. 10.14744/scie.2019.29290
2. Hernández-Martínez C, Espinosa-Rosales F, Espinosa-Padilla S, Hernandez-Martinez A, Blancas-Galicia L. Conceptos básicos de las inmunodeficiencias primarias Basics of primary immunodeficiencies. Rev Alerg Méx. 2016;63:180–189. 10.29262/ram.v63i2.146
3. Picard C, Gaspar HB, Al-Herz W, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2017;38:96–128. 10.1007/s10875-017-0464-9
4. Picard C, Al-herz W, Bousfiha A, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies expert committee for primary immunodeficiency 2015. J Clin Immunol. 2015;35:696–726. 10.1007/s10875-015-0201-1
5. González de la Calle V, Pérez-Andrés M, Puig Moron N. Inmunodeficiencias primarias. Medicine. 2016;12:1191–1200. 10.1016/j.med.2016.10.010
6. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies expert committee. J Clin Immunol. 2020;40:24–64.10. 10.1007/s10875-019-00737-x
7. Elsink K, van Montfrans JM, van Gijn ME. Cost and impact of early diagnosis in primary immunodeficiency disease: a literature review. Clin Immunol. 2020;213:108359. 10.1016/j.clim.2020.108359
8. Belén A, Oswaldo S. Análisis discriminante para predecir el diagnóstico clínico de inmunodeficiencias primarias: reporte preliminar. Rev Alerg Mex. 2015;62:125–133. 10.29262/ram.v62i2.66
9. Modell V, Orange JS, Quinn J, Modell F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey modell centers network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res. 2018;66:367–380. 10.1007/s12026-018-8996-5
10. Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974–1979. J Clin Immunol. 1982;2:86–92. 10.1007/BF00916891
11. Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001–2007. J Clin Immunol. 2014;34:954–961. 10.1007/s10875-014-0102-8
12. Geller-Bernstein C, Etzioni A. Pediatric allergy and immunology in Israel. Pediatr Allergy Immunol. 2013;24:187–194. 10.1111/pai.12044
13. Ishimura M, Takada H, Doi T, et al. Nationwide survey of patients with primary immunodeficiency diseases in Japan. J Clin Immunol. 2011;31:968–976. 10.1007/s10875-011-9594-7
14. Gathmann B, Grimbacher B, Beauté J, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157: 3–11. 10.1111/j.1365-2249.2009.03954.x
15. Leiva LE, Bezrodnik L, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: proceedings of the second Latin American Society for Immunodeficiencies (LASID) advisory board. Allergol Immunopathol. 2011;39:106–110. 10.1016/j.aller.2010.10.007
16. Fundación Diana Garcia de Olarte-FIP. Las inmunodeficiencias primarias o IDP. [cited 2020. Available at: http://fundacion-fip.org.
17. ESID Registry Working Party. ESID Registry—Working definitions for clinical diagnosis of IEI. 1–33 (2019).
18. McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. 2018;14:61. 10.1186/s13223-018-0290-5
19. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary immunodeficiency and autoimmunity: a comprehensive review. J Autoimmun. 2019;99:52–72. 10.1016/j.jaut.2019.01.011
20. Pedraza Á, Vargas-Rumilla MI, Ramírez-Roa JL. Registry of primary immunodeficiencies in children at a fourth level hospital. Bogota, 2010–2016. Rev Alerg Mex. 2016;65, 341–348. 10.29262/ram.v65i4.338
21. Riaño-Cardozo LR, Correa-Vargas N, Gallón-Duque A, Orrego JC, Franco JL. Reporte epidemiológico de inmunodeficiencias primarias en el Centro Jeffrey Modell de Colombia: 1987-2017. Rev Alerg Méx. 2018;65:139–140.
22. Chapel H, Prevot J, Gaspar HB, et al. Primary immune deficiencies—principles of care. Front Immunol. 2014;5:627. 10.3389/fimmu.2014.00627
23. Felgentreff K, Perez-Becker R, Speckmann C, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141:73–82. 10.1021/bi010196v
24. Knerr V, Grimbacher B. Primary immunodeficiency registries. Curr Opin Allergy Clin Immunol. 2007;7: 475–480. 10.1097/ACI.0b013e3282f2162c
25. Fernández F, Campillay R, Palma V, Norambuena X, Quezada A, Inostroza J. Deficiencia de anticuerpos específicos:-inmunodeficiencia primaria asociada a alergia respiratoria Specific antibody deficiency: Primary immunodeficiency associated to respiratory allergy. Rev Chil Pediatr. 2017;88: 252–257. 10.1016/j.rchipe.2016.08.006
26. Nonas S. Pulmonary manifestations of primary immunodeficiency disorders. Immunol Allergy Clin North Am. 2015;35:753–766. 10.1016/j.iac.2015.07.004
27. Modell V, Gee B, Lewis DB, et al. Global study of primary immunodeficiency diseases (PI)-diagnosis, treatment, and economic impact: An updated report from the Jeffrey Modell Foundation. Immunol Res. 2011;51:61–70. 10.1007/s12026-011-8241-y