
Download
ORIGINAL ARTICLE
Dedicator of cytokinesis protein 2 knockdown ameliorates LPS-induced acute lung injury by modulating Rac1/2 activity
Yan Zhoua, Wan Hub*
aDepartment of Respiratory Medicine, Third Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China
bDepartment of Respiratory and Critical Care Medicine, Zhuhai People’s Hospital, Zhuhai, Guangdong, P.R. China
Abstract
Dedicator of cytokinesis protein 2 (DOCK2) is a member of the cytoskeletal dynamics protein family, and is ubiquitously expressed in hematopoietic cells according to previous studies. This paper was intended to explore the underlying mechanism that DOCK2 might involve in the progression of acute lung injury (ALI). Following lipopolysaccharide (LPS) induction in the purchased A549 cells, the expression level of DOCK2 was determined and its knockdown was performed by transfection. Subsequently, the viability, inflammation, oxidative stress barrier, and apoptosis of transfected A549 cells were measured to observe the alterations. Inflammation-related and apoptosis-related proteins were measured by western blot analysis. Finally, 8-Chlorophenylthio-cyclic monophosphate (8-CPT), ras-related C3 botulinum toxin substrate (Rac) 1 agonist, was applied to treat cells for investigating the underlying mechanism regarding the role of DOCK2. According to the results, DOCK2 was upregulated in LPS-induced A549 cells. Following the knockdown of DOCK2, the release of inflammatory cytokines was alleviated, accompanied by attenuated oxidative stress, barrier injury, and apoptosis of LPS-induced A549 cells. Nonetheless, this trend was reversed by further treatment of 8-CPT. In summary, DOCK2 knockdown alleviates inflammation, oxidative stress, barrier injury, and apoptosis of LPS-induced A549 cells by associating with Rac1/Rac2. These findings highlighted the therapeutic potential of DOCK2 for the treatment of ALI.
Key words: acute lung injury, apoptosis, dedicator of cytokinesis protein 2, inflammation response, ras-related C3 botulinum toxin substrate 1/2
*Corresponding author: Wan Hu, Department of Respiratory and Critical Care Medicine, Zhuhai People’s Hospital, Zhuhai, Guangdong, 519000, P.R. China. Email address: [email protected]
Received 17 November 2021; Accepted 21 December 2021; Available online 1 May 2022
Zhou Y and Hu W
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Acute lung injury (ALI) constitutes a major health problem featured by the acute onset of hypoxemia related to increased pulmonary vascular permeability.1 The advances in diagnostic techniques and treatment approaches of ALI cannot substantially decrease the mortality of ALI, which has high levels of 25% to 50%.2,3 Keen interest of scholars and experts in developing new biomarkers for the treatment of ALI in recent decades has brought about exponentially increasing studies, and it is acknowledged that the identification of these biomarkers will help predict and improve the clinical outcome of patients with ALI and related diseases.4
Dedicator of cytokinesis protein 2 (DOCK2) is a member of the cytoskeletal dynamics protein family, and is ubiquitously expressed in hematopoietic cells.5 It functions as a critical ras-related C3 botulinum toxin substrate (Rac) regulator that can affect cellular activities, including migration, activation, and proliferation of lymphocytes.6 Patients with mutated DOCK2 show great potential to present with lymphopenia and combined immunodeficiency diseases.7 Furthermore, the expression of LEF-1, a negative regulator of CD21, was increased in these patients, which can affect the cell metabolism.8 Studies have shown that small molecule inhibitors of DOCK2 can effectively block the inflammatory response.9 At the same time, the expression of DOCK2 is upregulated in myocarditis, and LPS-induced cardiomyocytes can also upregulate the expression of DOCK2, the inhibition of which can effectively block the apoptosis and inflammation of cardiomyocytes.10
Moreover, some literature suggests that DOCK2, which is upregulated in ALI, can be identified as a potential lung injury marker.11 However, it remains elusive regarding the role of DOCK2 in ALI, and DOCK2 is also an atypical Rac activator.12 Inhibition of Rac activity can effectively alleviate the severe symptoms in LPS-induced ALI mice model.13 Therefore, conjecture that DOCK2 knockdown may inhibit the progression of ALI and its underlying mechanism has been put forward. Same as the previous studies establishing in vitro ALI models, LPS was used herein for the induction of ALI.
Materials and methods
Cell culture and transfection
Human nonsmall cell lung cancer cells A549 (STR Authentication; Shanghai YBio Biological Technology Co., Ltd.) were recovered in a 37°C water bath, suspended with RPMI 1640, supplemented with 5% heat-inactivated fetal bovine serum and streptomycin, centrifuged, resuspended, and seeded. The cells were then cultured in the aforementioned complete medium at 37°C and 5% CO2 in humidified air. The cells after incubation for 2 days were exposed to LPS for 24 h. DOCK2 siRNAs (si-DOCK2-1/2) and scrambled siRNA as a negative control (si-NC) were obtained from Gene Pharma Corporation (Shanghai, China) and transfected into the cells using Lipofectamine@ RNAi Max (Invitrogen, Carlsbad, CA). Following transfection for 6 h, the reagent in the petri dish was replaced with fresh complete medium, and the cells were cultured for another 2 days. Afterward, cells were digested by 0.25% trypsin (Beyotime, China), and collected for the subsequent assays. 8-CPT (5 µM), Rac1 agonist, was purchased from Sigma-Aldrich and applied to activate Rac1 in the LPS-induced A549 cells.
Quantitative Real-time PCR
Total RNA was extracted from A549 cells using TRIzol reagent (Invitrogen, CA, USA). Prime ScriptTM RT Master Mix (Takara, Otsu, Japan) was conducted for the RNA reverse transcription from mRNA into cDNA. Quantitative Real-time PCR (RT-qPCR) was performed by SYBR Green master mix (catalog number QP005; Genecopoeia). The available primers sequences were as follows: DOCK2-F: 5'-CTTGGAGGTCCTCAGCTGTC-3', DOCK2-R: 5'-GTCTGAGCT GGTCTGGAAGG-3'; GAPDH-F: 5'-AGGTCGGTGTGAACGGA TTTG-3', GAPDH-R: 5'-TGTAGACCATGTAGTTGAGGTCA-3'. Applied Biosystems 7900 Real-Time PCR System (Applied Biosystems, Foster City, CA) was used for RNA quantification assay. GAPDH were used as an internal control of mRNAs and fold change was determined by the 2-∆∆Ct method.
CCK-8 assay
A549 cells were inoculated in 96-well plates at a density of 3 × 103 cells/well. Each group of cells was collected at 48 h and then 10 µL CCK-8 solution was added to the medium. Cells were maintained to be incubated at 37°C for another 4 h. Absorbance was measured at 450 nm by a microplate reader (Bio-RadLabs, Sunnyvale, CA).
Enzyme-linked immunosorbent assay
Cell supernatant sample was centrifuged at 500 × g at 4°C for 5 min and the supernatant was collected. The expression levels of tumor necrosis factor (TNF)-α (cat. no. H052-1), Interleukin (IL)-6 (cat. no. H007-1), IL-1β (cat. no. H002), and PGE2 (cat. no. H099-1; Nanjing JianCheng Bioengineering Institute) were measured by the corresponding ELISA kit according to the manufacturer’s recommendations. The absorbance was determined at 450 nm by a microplate reader (BioTek Instruments, Inc., USA).
Determination of oxidative stress indicators
A549 cells (1 × 106) were lysed using radio-immunoprecipitation assay (RIPA) lysis buffer, the lysate was centrifuged at 10,000 × g at 4°C for 10 min, and the supernatant was collected. Malondialdehyde (MDA; cat. no. A003-1-1), SOD (cat. no. A001-1), and GSH-Px (cat. no. A005-1; Nanjing JianCheng Bioengineering Institute) assay kits were used to assess the content of the corresponding function indicators, according to the manufacturer’s instructions.
Transepithelial electrical resistance assay
A549 cells (3 × 104) were seeded into the upper chamber of 24-well Transwell system plates (pore size, 0.4 µm; Corning, Inc.) and incubated with RPMI 1640 for 20 days at 37°C. Transepithelial electrical resistance (TEER) was measured daily using an epithelial volt ohmmeter. The calculation used was as follows: TEER = (R1–R2) × M. R1 represents the background resistance, R2 represents the collagen layer and membrane insert resistance, and M represents the insert membrane area.
TUNEL assay
A549 cells (5 × 105/well) were seeded into a 24-well plate and cultured until they reached ∼80% confluence. Cells were fixed with 4% paraformaldehyde for 30 min at room temperature and permeated with phosphate buffer saline (PBS) containing 0.3% Triton X-100 for another 5 min at room temperature. Apoptosis was detected and quantified by TUNEL assay using the One Step TUNEL Apoptosis Assay Kit (cat. no. C1088; Beyotime, China) in accordance with the manufacturer’s protocol. The apoptotic rate was expressed as a percentage of TUNEL-positive cells.
Western blotting analysis
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors for 30 min. Protein concentrations were determined by a BCA protein assay kit (Beyotime, China). The samples were separated by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes, which were then blocked with 5% skim milk for 2 h and incubated with the primary antibodies overnight at 4°C. Primary antibodies were used as follows: anti-DOCK2 (cat. no. ab124838; 1:1,000; Abcam), anti-TNF-α (cat. no. 8184S, 1:1,000; Cell Signaling Technology), anti-IL-6 (cat. no. 12153S; 1:1,000; Cell Signaling Technology), anti-Bcl-2 (cat. no. ab32124; 1:1,000; Abcam), anti-Bax (cat. no. ab32503; 1:1,000; Abcam), anti-caspase-3 (cat. no. Cayman_10010209; 1:1,000; Zen BioScience), occludin (cat. no. ab216327; 1:1,000; Abcam), claudin 1 (cat. no. ab211737; 1:2,000; Abcam), ZO-1 tight junction protein (ZO-1; cat. no. ab216880; 1:1,000; Abcam), and anti-Rac1/2 (cat. no. 2465S; 1:1,000; Abcam). Following that, the membranes were incubated with HRP-conjugated secondary antibody (cat. no. ab97080; 1:5,000; Abcam) at room temperature for 1 h. Then, the bands were incubated in enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ), and intensities were quantified by Image J analysis software (v1.8, National Institutes of Health).
Statistical analysis
Statistical data are expressed as mean ± SD. Differences between the control and the LPS groups were analyzed by using the unpaired Student’s t-test, while those among multiple groups were analyzed by using the one-way ANOVA and Tukey’s post hoc test. P values less than 0.05 were considered significant. Analysis for significance was conducted by GraphPad Prism 8.0 (GraphPad Software Inc.).
Results
The impact of DOCK2 on the cell viability of LPS-induced A549 cells
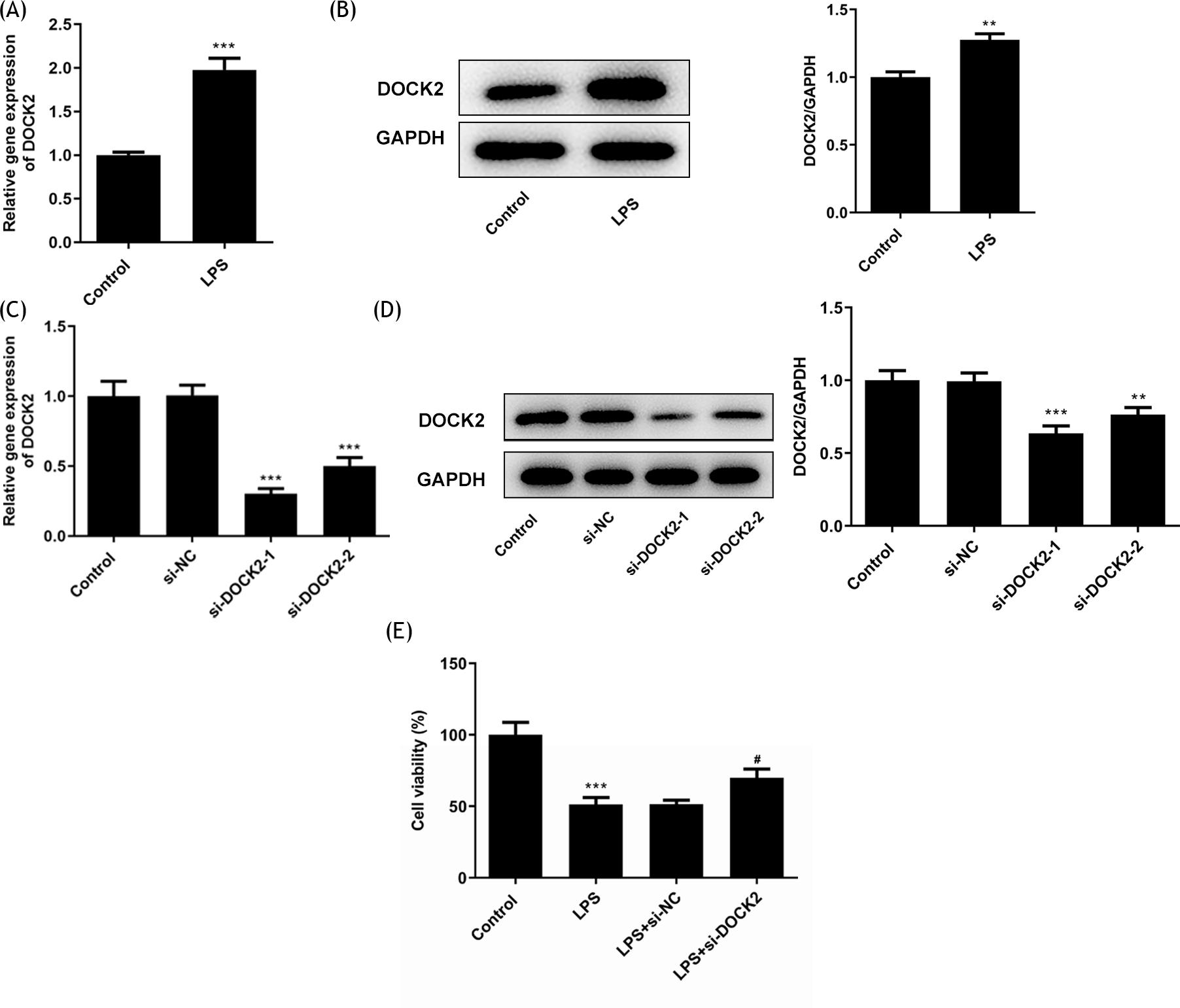
To assess the impact of DOCK2 on the viability of ALI cells, the expression level of DOCK2 was detected in LPS-induced A549 cells via RT-qPCR and western blotting. In comparison with the control group, the expression of DOCK2 in the cell model was evidently elevated (Figure 1A and B). As DOCK2 was up-regulated in the models, its expression was knocked down in order to study its regulatory effect on cells. DOCK2 knockdown was performed by transfecting with si-DOCK2-1 and si-DOCK2-2 into A549 cells, and si-DOCK2-2 was selected for the following experiments due to its excellent transfection efficiency from the results of RT-qPCR and western blotting (Figure 1C and D). The cell viability of A549 cells upon LPS induction examined via a CCK8 assay was reduced to one half of that in the control group, while DOCK2 knockdown partially reversed this trend by upregulating the damaged cell viability (Figure 1E).
Figure 1 The impact of DOCK2 on the cell viability of LPS-induced A549 cells. The expression level of DOCK2 in LPS-induced A549 cells was determined using (A) RT-qPCR and (B) western blotting. The expression levels of DOCK2 in each group of A549 cells were determined using (C) RT-qPCR and (D) western blotting. (E) The cell viability of each group of A549 cells was determined using a CCK8 assay. **p˂0.01 and ***p˂0.001 versus control; #p˂0.05 versus LPS + si-NC group.
DOCK2 knockdown alleviates inflammation, oxidative stress, barrier dysfunction, and apoptosis of LPS-induced A549 cells
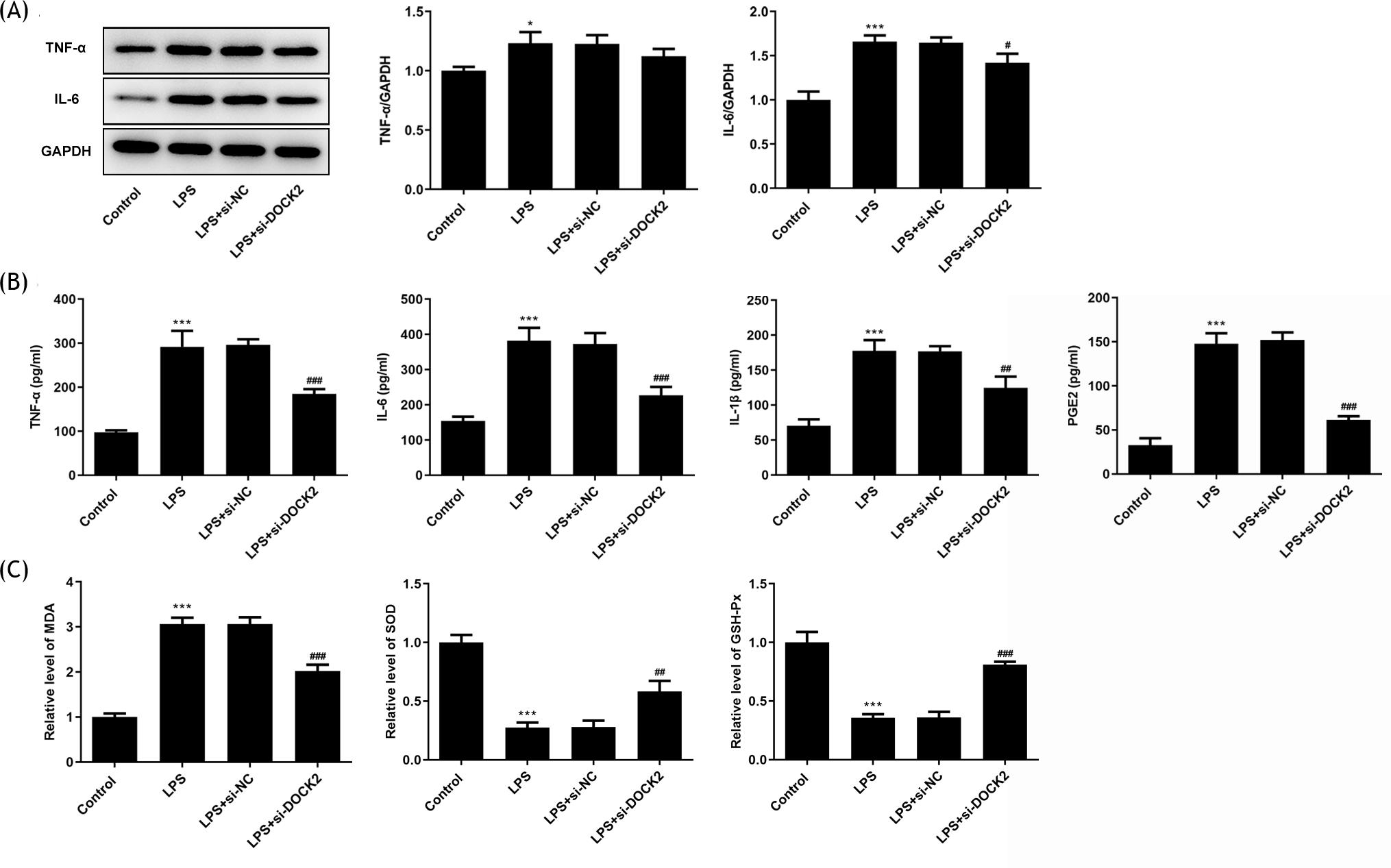
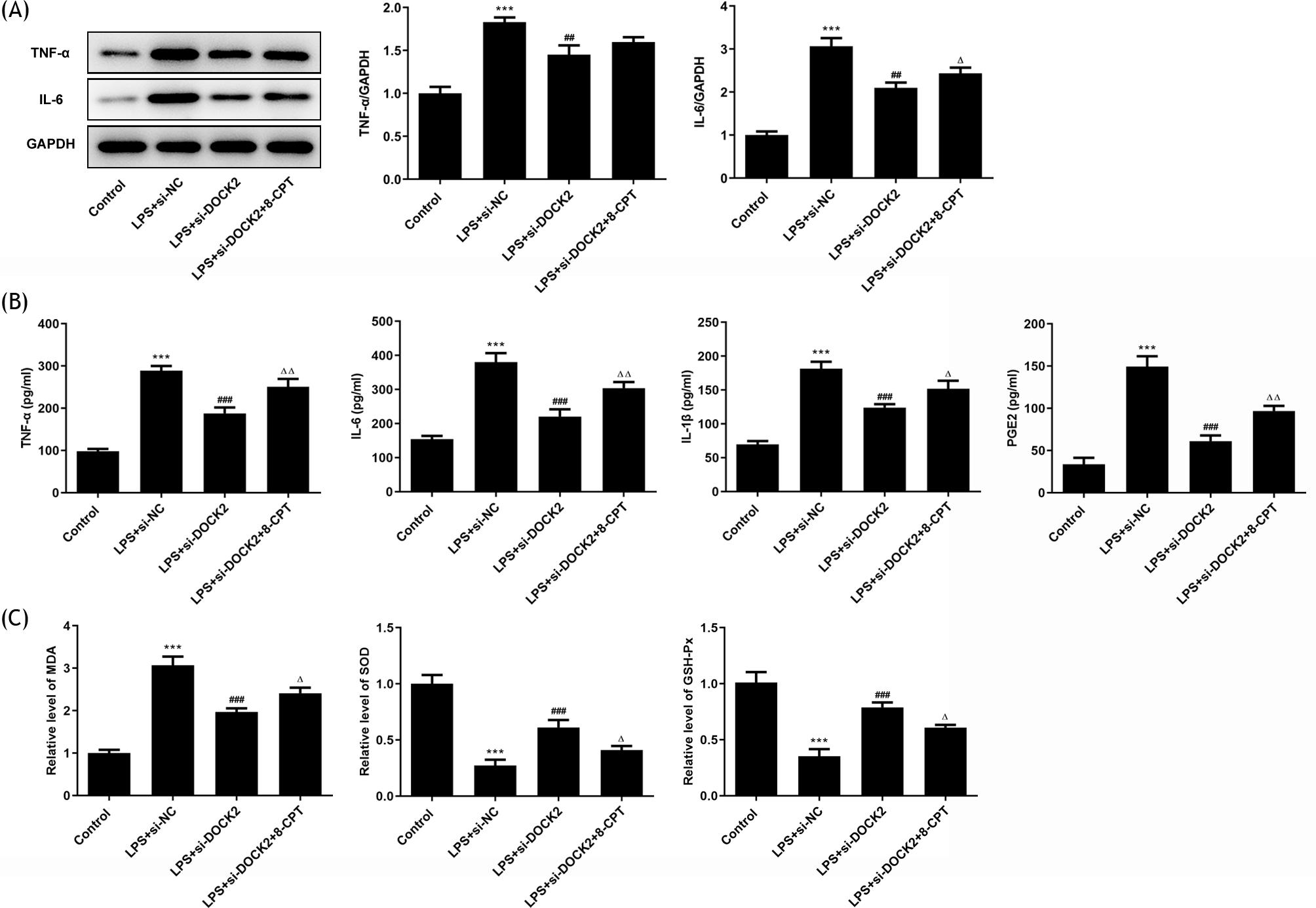
As inflammation and apoptosis are hallmarks of ALI, the inflammation and apoptosis were measured in LPS-induced A549 cells. From the results of western blotting, the expression of inflammation-related proteins, TNF-α and IL-6, was elevated in A549 cells upon LPS stimulation, whereas DOCK2 knockdown inhibited their expression (Figure 2A). Consistently, ELISA assay results demonstrated that the released levels of TNF-α, IL-6, IL-1β, and PGE2▫ in cells transfected with si-DOCK2 were all declined compared with si-NC groups (Figure 2B). Afterward, the levels of MDA, SOD, and GSH-Px in each group were determined using assay kits. The level of MDA was decreased while the levels of SOD and GSH-Px were increased in the si-DOCK2 groups compared to the si-NC groups (Figure 2C).
Figure 2 DOCK2 knockdown alleviates inflammation and oxidative stress of LPS-induced A549 cells. (A) The expression levels of TNF-α and IL-6 in each group of A549 cells were detected using western blotting. (B) The content of TNF-α, IL-6, IL-1β, and PGE2 in each group of A549 cells were detected using ELISA assay. (C) The levels of MDA, SOD, and GSH-Px were determined using assay kits. *p˂0.05 and ***p˂0.001 versus control; #p˂0.05, ##p˂0.01, and ###p˂0.001 versus LPS + si-NC group.
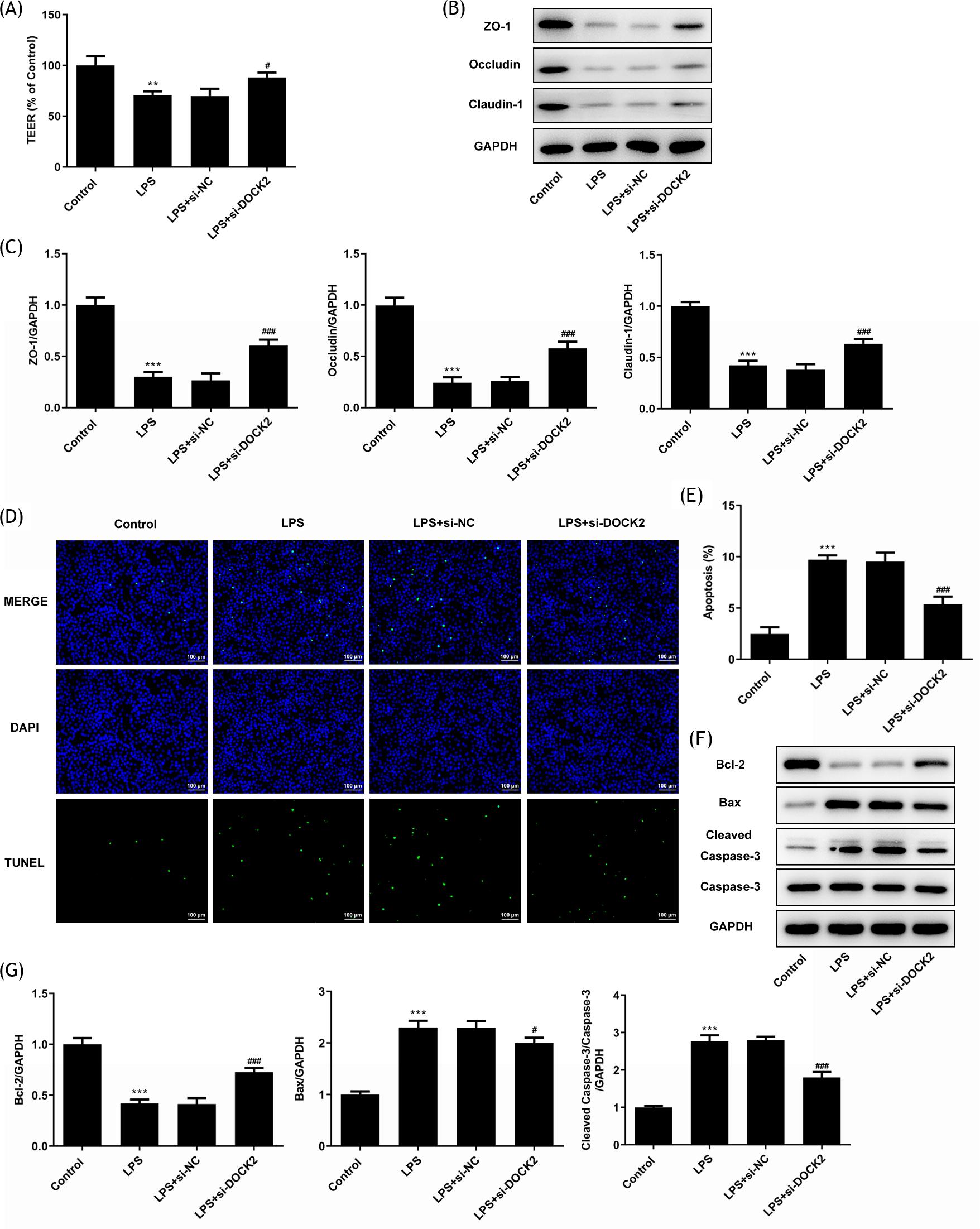
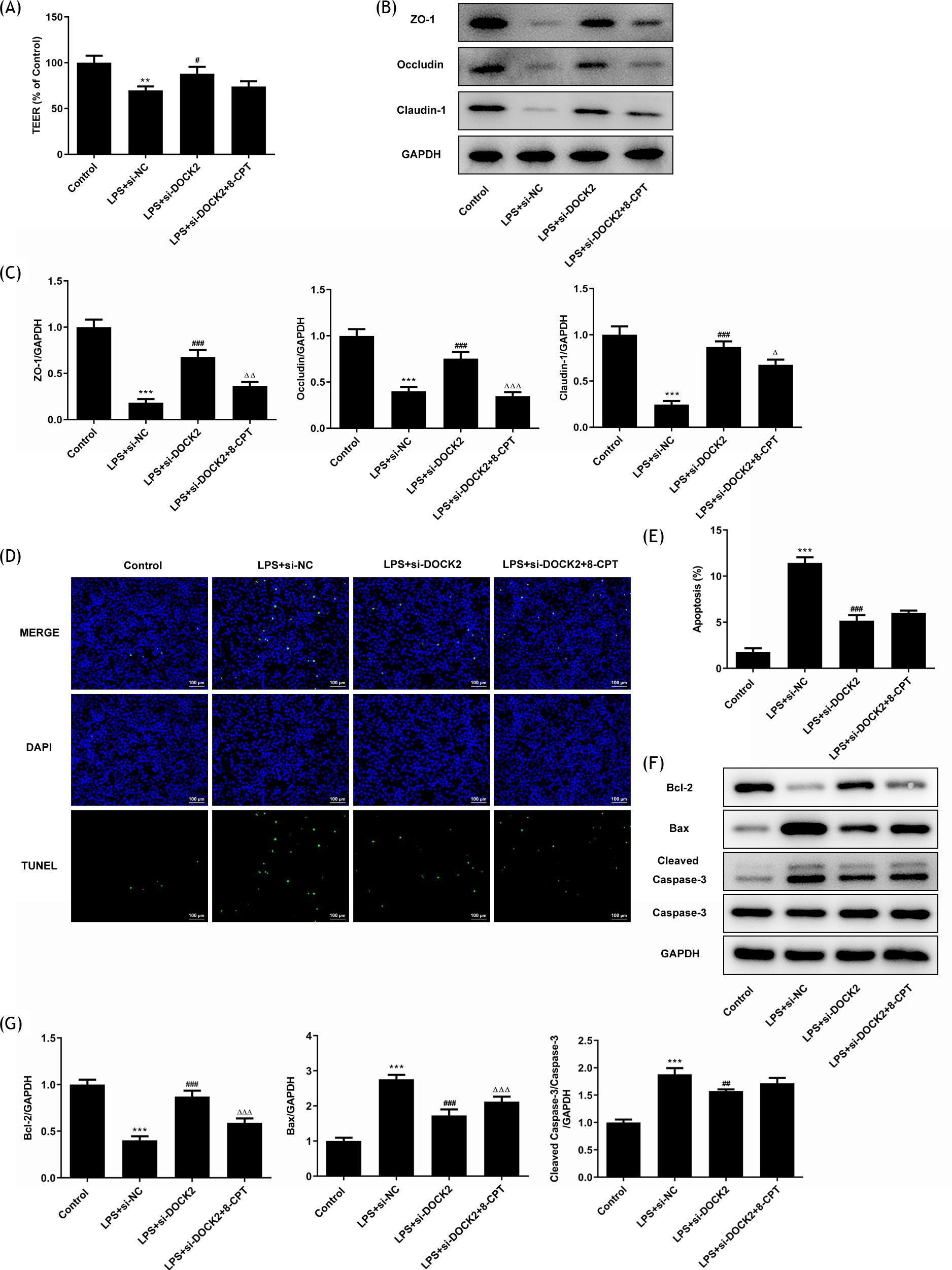
In addition, cell monolayer permeability was measured by TEER measurement. The result indicated that the LPS declined the TEER value and DOCK2 knockdown reversed this value (Figure 3A). Subsequently, the protein expression levels of ZO-1, Occludin, and Claudin-1 were also determined. The western blot results revealed that these protein expression levels were all downregulated in the LPS group, and DOCK2 knockdown reversed the decline (Figure 3B and C). Moreover, the results of TUNEL assay indicated that DOCK2 knockdown decreased the apoptosis rate of A549 cells stimulated by LPS (Figure 3D and E), accompanied by upregulated expression of Bcl-2 and downregulated expression of Bax and cleaved caspase 3, displayed via the results of western blotting (Figure 3F and G).
Figure 3 DOCK2 knockdown alleviates barrier dysfunction and apoptosis of LPS-induced A549 cells. (A) The levels of barrier function were determined using the TEER assay. (B and C) The expression levels of ZO-1, Occludin, and Claudin-1 in each group of A549 cells were examined using western blotting. (D and E) The apoptosis rates of each group of A549 cells were determined using TUNEL assay. (F and G) The expression levels of Bcl-2, Bax, cleaved caspase 3 and caspase 3 in each group of A549 cells were examined using western blotting. ***p˂0.001 versus control; #p˂0.05 and ###p˂0.001 versus LPS + si-NC group.
The impact of DOCK2 on the activities of Rac1/Rac2
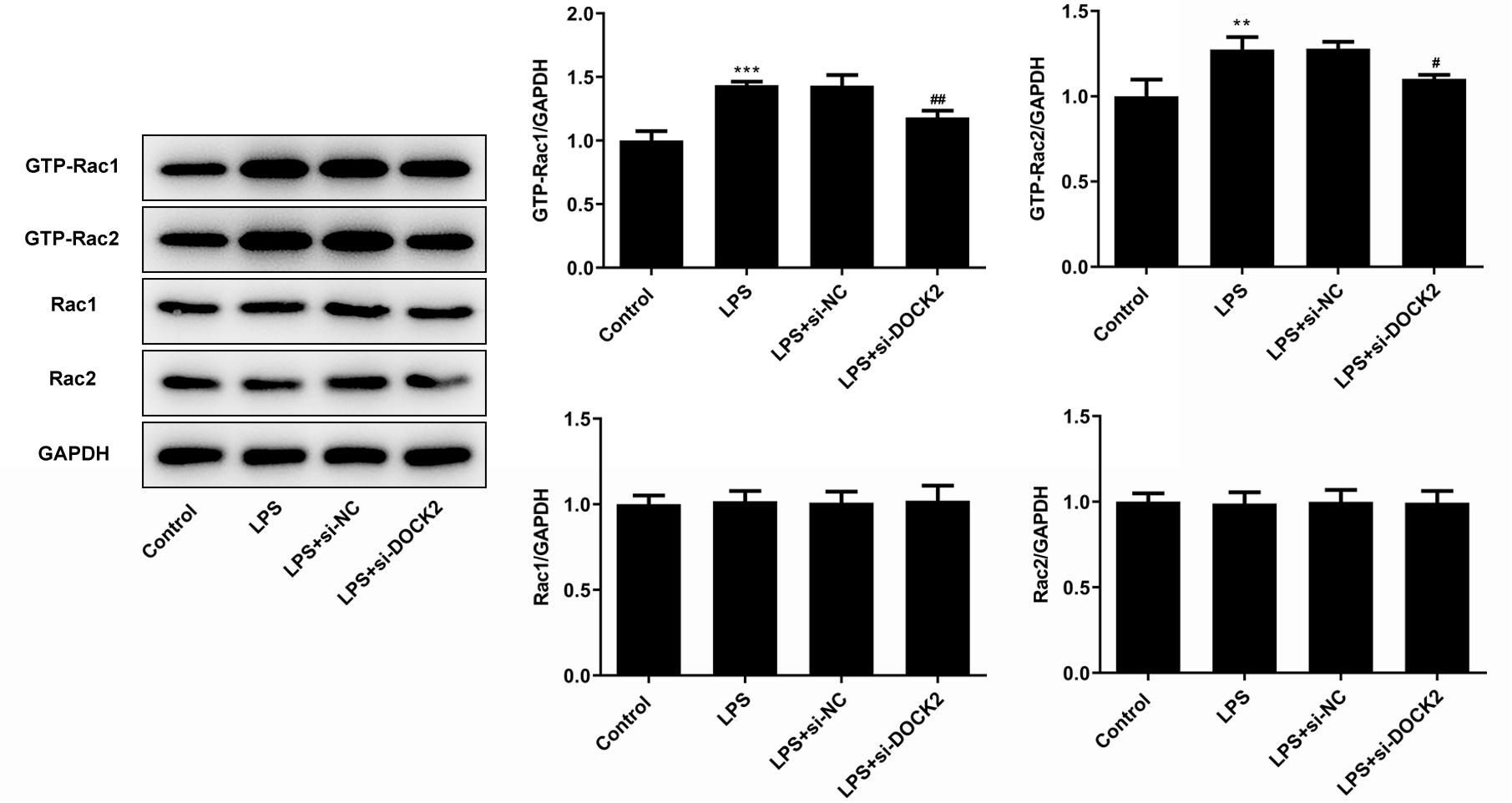
The influence of deficiency of DOCK2 expression on the activities of Rac1/Rac2 was explored. By western blot analysis, LPS activated the expression of GTP-Rac1 and GTP-Rac2 (the activated morphology of Rac1 and Rac2) and DOCK2 knockdown inactivated their expression, demonstrating the positive relationship between DOCK2 expression and Rac1/Rac2 activities (Figure 4). Nevertheless, the expression of Rac1 and Rac2 remained unchanged during the whole process. Overall, the above results indicated that DOCK2 could positively regulate the expression of Rac1 and Rac2.
Figure 4 The impact of DOCK2 on the activities of Rac1/Rac2. The expression levels of Rac1/Rac2 in each group of cells were determined using western blotting. **p˂0.01 and ***p˂0.001 versus control; #p˂0.05 and ##p˂0.01 versus LPS + si-NC group.
DOCK2 knockdown alleviates inflammation, oxidative stress, barrier dysfunction, and apoptosis of LPS-induced A549 cells via Rac1/Rac2
To confirm whether DOCK2 has an impact on the cellular activities of LPS-induced A549 cells via Rac1/Rac2, western blotting and ELISA assay were performed. 5 µM of 8-CPT, a sort of Rac1 agonist, was applied to treat LPS-induced transfected cells. As expected, the expression of TNF-α and IL-6 in LPS-induced transfected cells was enhanced upon 8-CPT treatment (Figure 5A). Consistent result regarding TNF-α, IL-6, IL-1β, and PGE2 production was demonstrated from the determination of their contents (Figure 5B). And, 8-CPT treatment markedly reduced the levels of SOD and GSH-Px, whereas the MDA level was increased (Figure 5C).
Figure 5 DOCK2 knockdown alleviates inflammation and oxidative stress of LPS-induced A549 cells via Rac1/Rac2. (A) The expression levels of TNF-α and IL-6 in each group of A549 cells were detected using western blotting. (B) The content of TNF-α, IL-6, IL-1β, and PGE2 in each group of A549 cells was detected by ELISA assay. (C) The levels of MDA, SOD, and GSH-Px were determined using assay kits. ***p˂0.001 versus control; ##p˂0.01 and ###p˂0.001versus LPS + si-NC group; ∆p˂0.05 and ∆∆p˂0.01 versus LPS + si-DOCK2 group.
Moreover, 8-CPT was found to decrease the TEER value (Figure 6A), as well as the protein expression levels of ZO-1, Occludin, and Claudin-1 (Figure 6B and C). The apoptosis of LPS + si-DOCK2 + 8-CPT group was evidently higher than that of LPS + si-DOCK2 group (Figure 6D and E). Compared with the LPS + si-DOCK2 group, treatment with 8-CPT suppressed the anti-apoptotic Bcl-2 expression while it enhanced the pro-apoptotic Bax and cleaved caspase3 expression in LPS-induced transfected cells (Figure 6F and G). These results suggested that the knockdown of DOCK2 expression alleviates inflammation and apoptosis of LPS-induced A549 cells via Rac1/Rac2.
Figure 6 DOCK2 knockdown alleviates barrier dysfunction and apoptosis of LPS-induced A549 cells via Rac1/Rac2. (A) The levels of barrier function were determined using the TEER assay. (B and C) The expression levels of ZO-1, Occludin, and Claudin-1 in each group of A549 cells were examined using western blotting. (D and E) The apoptosis rates of each group of A549 cells were determined using TUNEL assay. (F and G) The expression levels of Bcl-2, Bax, cleaved caspase 3 and caspase 3 in each group of A549 cells were examined using western blotting. ***p˂0.001 versus control; ##p˂0.01 and ###p˂0.001versus LPS + si-NC group; ∆∆∆p˂0.001 versus LPS + si-DOCK2 group.
Discussion
In this study, LPS was used for the establishment of the ALI cell model to explore the underlying mechanism of DOCK2 in ALI progression. Overall, our current in vitro study demonstrated that the knockdown of DOCK2 played a beneficial role in the progression of ALI, including upregulated cell viability, alleviated inflammation, oxidative stress, barrier dysfunction, and apoptosis in LPS-induced A549 cells. In addition, decreased DOCK2 expression was associated with suppressed Rac1/Rac2 activities, which affected the abovementioned aspects in the cell model.
DOCK2 takes control of various immunological cell functions, such as helper T cell differentiation and type I interferon induction.14 The finding that mice with DOCK2 knockout was smaller in size compared to mice without gene mutations led scholars to put forward the conjecture that DOCK2 might control the metabolism of mice.15 It was previously reported that depletion of DOCK2 attenuated adipose tissue and systemic inflammation by reducing the release of inflammatory factors including leptin, IL-6, IL-10, TNF-α, and IL-12 in adipose tissue.15 In the present study, in addition to exploring the role of DOCK2 knockdown in suppressing the inflammation, the effects of alleviating oxidative stress, barrier dysfunction were also found in LPS-induced A549 cells. As apoptosis is also essential for the progression of ALI,16 the apoptosis of LPS-induced A549 cells transfected with si-DOCK2 was determined herein. Notably, DOCK2 knockdown was found to lead to attenuated apoptosis rate of LPS-induced A549 cells. This is not the first time that DOCK2 knockdown has been found to inhibit cell apoptosis. As aforementioned, it has also been proven to inhibit cardiomyocyte apoptosis. These findings expand the scope of understanding of DOCK2 and indicate that DOCK2 may be an effective target to alleviate various tissue damages.
Afterward, we have confirmed the correlation of DOCK2 with Rac1/Rac2 in the progression of ALI. Rac1 and Rac2 are involved in a wide range of physiological processes and the immune system in cells.17 Rac1 takes typical accountability for the actin polymerization, which contributes to the formation of lamellipodia and regulation of adhesion formation.18 Recent evidence has indicated that DOCK2 can activate Rac and regulate actin cytoskeleton by interacting with ELMO1.19 Furthermore, a previous report has noted that the expression of Rac1 and Rac2 was dramatically reduced upon B cell receptor (BCR) excitation in B cells where DOCK2 absent.20 Both studies reveal that there is a certain association between DOCK2 and Rac signals. Consistent with these lines of evidence, we found that knockdown of DOCK2 in LPS-treated A549 cells led to inactivated Rac1/Rac2 activities, suggesting a positive relationship between DOCK2 and Rac1/Rac2 activities in ALI. Hence, we believe that the Rac signal plays an essential role in alleviating ALI. Certainly, this signal has also been found to be involved in blood–brain barrier21 and vascular endothelial dysfunctions.22 More research on this signal is considered valuable for the future.
Conclusion
In summary, our current study demonstrated that DOCK2 was elevated in LPS-induced A549 cells. Furthermore, DOCK2 knockdown alleviates inflammation, oxidative stress, barrier dysfunction, and apoptosis of LPS-induced A549 cells via Rac1/Rac2. These findings highlighted the potential of DOCK2 as a biomarker and therapeutic target to serve patients suffering from ALI. However, this article is limited to an in vitro study, and more in vivo studies are required for verification in the future.
REFERENCES
1. Quinlan GJ, Evans TW. Acute respiratory distress syndrome in adults. Hosp Med. 2000;61(8):561–563. 10.12968/hosp.2000.61.8.1400
2. McIntyre RC Jr., Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Crit Care Med. 2000;28(9):3314–3331. 10.1097/00003246-200009000-00034
3. Blank R, Napolitano LM. Epidemiology of ARDS and ALI. Crit Care Clin. 2011;27(3):439–458. 10.1016/j.ccc.2011.05.005
4. Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: Prognostic and pathogenetic significance. Am J Respir Crit Care Med. 1997;155(4):1187–1205. 10.1164/ajrccm.155.4.9105054
5. Reif K, Cyster J. The CDM protein DOCK2 in lymphocyte migration. Trends Cell Biol. 2002;12(8):368–373. 10.1016/S0962-8924(02)02330-9
6. Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res. 2013;319(15):2343–2349. 10.1016/j.yexcr.2013.07.024
7. Alosaimi MF, Shendi H, Beano A, Stafstrom K, El Hawary R, Meshaal S, et al. T-cell mitochondrial dysfunction and lymphopenia in DOCK2-deficient patients. J Allergy Clin Immunol. 2019;144(1):306–9 e2. 10.1016/j.jaci.2019.02.020
8. Yang L, Jing Y, Wang W, Ying W, Lin L, Chang J, et al. DOCK2 couples with LEF-1 to regulate B cell metabolism and memory response. Biochem Biophys Res Commun. 2020;529(2):296–302. 10.1016/j.bbrc.2020.05.152
9. Nishikimi A, Uruno T, Duan X, Cao Q, Okamura Y, Saitoh T, et al. Blockade of inflammatory responses by a small-molecule inhibitor of the Rac activator DOCK2. Chem Biol. 2012;19(4):488–497. 10.1016/j.chembiol.2012.03.008
10. Wang L, Zhang Y, Zhu G, Ma Y, Zuo H, Tian X. miR-16 exhibits protective function in LPS-treated cardiomyocytes by targeting DOCK2 to repress cell apoptosis and exert anti-inflammatory effect. Cell Biol Int. 2020;44(8):1760–1768. 10.1002/cbin.11371
11. Xu X, Zhu Q, Zhang R, Wang Y, Niu F, Wang W, et al. ITRAQ-based proteomics analysis of acute lung injury induced by oleic acid in mice. Cell Physiol Biochem. 2017;44(5):1949–1964. 10.1159/000485885
12. Terasawa M, Uruno T, Mori S, Kukimoto-Niino M, Nishikimi A, Sanematsu F, et al. Dimerization of DOCK2 is essential for DOCK2-mediated Rac activation and lymphocyte migration. PLoS One. 2012;7(9):e46277. 10.1371/journal.pone.0046277
13. Yao HY, Chen L, Xu C, Wang J, Chen J, Xie QM, et al. Inhibition of Rac activity alleviates lipopolysaccharide-induced acute pulmonary injury in mice. Biochim Biophys Acta. 2011;1810(7):666–674. 10.1016/j.bbagen.2011.03.020
14. Guo X, Chen SY. Dedicator of cytokinesis 2 in cell signaling regulation and disease development. J Cell Physiol. 2017;232(8):1931–1940. 10.1002/jcp.25512
15. Guo X, Li F, Xu Z, Yin A, Yin H, Li C, et al. DOCK2 deficiency mitigates HFD-induced obesity by reducing adipose tissue inflammation and increasing energy expenditure. J Lipid Res. 2017;58(9):1777–1784. 10.1194/jlr.M073049
16. Li S, Wei P, Zhang B, Chen K, Shi G, Zhang Z, et al. Apoptosis of lung cells regulated by mitochondrial signal pathway in crotonaldehyde-induced lung injury. Environ Toxicol. 2020;35(11):1260–1273. 10.1002/tox.22991
17. Fenteany G, Glogauer M. Cytoskeletal remodeling in leukocyte function. Curr Opin Hematol. 2004;11(1):15–24. 10.1097/00062752-200401000-00004
18. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81 (1):53–62. 10.1016/0092-8674(95)90370-4
19. Sanui T, Inayoshi A, Noda M, Iwata E, Stein JV, Sasazuki T, et al. DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood. 2003;102(8):2948–2950. 10.1182/blood-2003-01-0173
20. Ushijima M, Uruno T, Nishikimi A, Sanematsu F, Kamikaseda Y, Kunimura K, et al. The Rac activator DOCK2 mediates plasma cell differentiation and IgG antibody production. Front Immunol. 2018;9:243. 10.3389/fimmu.2018.00243
21. Zhai L, Liu M, Wang T, Zhang H, Li S, Guo Y. Picroside II protects the blood-brain barrier by inhibiting the oxidative signaling pathway in cerebral ischemia-reperfusion injury. PLoS One. 2017;12(4):e0174414. 10.1371/journal.pone.0174414
22. Kunimura K, Miki S, Takashima M, Suzuki JI. S-1-propenylcysteine improves TNF-α-induced vascular endothelial barrier dysfunction by suppressing the GEF-H1/RhoA/Rac pathway. Cell Commun Signaling. 2021;19(1):17. 10.1186/s12964-020-00692-w