
Download
ORIGINAL ARTICLE
Experience of Autoimmune and autoinflammatory diseases in a Turkish pediatric cohort with primary immunodeficiencies
Dilek Özcana*, Mahir Serbesa, Rabia Miray Kışla Ekincib, Sibel Balcı Çetinkayab, Ayşe Şenay Şaşihüseyinoğlua, Atıl Bişginc, Derya Ufuk Altıntaşa
aDepartment of Pediatric Allergy-Immunology, Faculty of Medicine, Çukurova University, Adana, Turkey
bDepartment of Pediatric Rheumatology, Faculty of Medicine, Çukurova University, Adana, Turkey
cDepartment of Medical Genetics, Faculty of Medicine, Çukurova University, Adana, Turkey
Abstract
Background Primary immunodeficiency diseases (PID) are the diseases characterized by a dysfunction of the immune system. Affected patients share a different phenotype such as chronic infections, allergy, autoimmunity, and autoinflammation.
Methods In all, 433 children with PID were enrolled in this study. Clinical, laboratory, and demographic data of patients were reviewed retrospectively to investigate autoimmune and autoinflammatory complications. Autoinflammation in all patients with inflammation was confirmed by genetic analysis after excluding infectious etiology.
Results Clinical features of 433 PID patients were evaluated retrospectively with long-term follow-up. Autoimmune disorders were identified in 69 (15.9%) patients with PID; 31 (45%) patients had a history of autoimmune disease before diagnosis of PID. The frequency of autoimmunity in immune dysregulation subgroup (76.6%) was higher than other forms of PID. The most common autoimmune manifestations were reported to be Addison’s disease, hypoparathyroidism, and autoimmune hemolytic anemia. Autoinflammation were identified in 22 of the 433 (5.1%) patients with PID, including hyper immunoglobulin D syndrome (n = 9), Aicardi–Goutieres syndrome 1 (n = 6), adenosine deaminase 2 deficiency (n = 3), Blau syndrome (n = 2), tumor necrosis factor (TNF) receptor-associated periodic syndrome (n = 1), and auto-inflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation syndrome (n = 1).
Conclusions It is important to recognize association between autoimmunity, autoinflammation, and PID, which in the future could be useful for increased awareness and early diagnosis for these diseases.
Key words: autoimmunity, autoinflammation, immune dysregulation, immune system, primary immune diseases
Corresponding author: Dilek Özcan, Department of Pediatric Allergy-Immunology, Faculty of Medicine, Çukurova University, Adana, Turkey. E-mail address: [email protected]
Received 23 May 2021; Accepted 5 July 2021; Available online 1 November 2021
Copyright: Özcan D, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Primary immunodeficiency diseases (PID) are characterized by clinical symptoms ranging from recurrent infections to autoimmune diseases. Although two paradoxical responses of the immune system are autoimmunity and immunodeficiency, defects in the mechanisms of immunity may simultaneously result in these manifestations.1 Autoimmune and autoinflammatory dysregulation are frequent characteristics of primary immunodeficiencies. In addition, autoimmune manifestations may be the first sign of PID in patients with no considerable history of severe or recurrent infections.2 Autoimmune manifestations are very heterogeneous, and include hematologic, rheumatologic, dermatologic, and gastrointestinal diseases.3
Development of autoimmunity in patients with PID does not remain fully elucidated but is thought to be multifactorial. Both genetic and environmental interactions have been hypothesized to play a role in pathogenesis. Studies suggest that this could be due to increased autoreactive B cells and deficiency of T cell regulation. Additionally, innate immune system and dysfunctional T cell reactivation because of chronic infection has been reported to be included in the relationship between PID and autoimmunity. Another mechanism involved in the development of autoimmunity could be the inability to clear apoptotic cells or immunocomplexes.4 It is also increasingly evident that autoinflammatory manifestations are frequently seen as complications of primary immunodeficiency disorders.
The aim of our study was to define the clinical and laboratory characteristics of autoimmune and autoinflammatory manifestations in order to identify these as remarkable signs in the early diagnosis of PID.
Materials and Methods
Patients
In this study, we reviewed the medical records of patients with PIDs referred to Çukurova University Medical School Hospital between 2000 and 2020. All the patients included in the study were living in the southern region of Turkey. Diagnosis of PID was ensured according to the diagnostic criteria of the European Society for Immunodeficiencies and/or positive gene screening results.5 Patients were also classified into different categories of PIDs in accordance with the Primary Immunodeficiency Diseases Committee Report of the International Union of Immunological Societies 2019.6 Gender of patients, age at diagnosis, age at initial time of symptoms associated with autoimmunity, current age, genetic mutations, laboratory findings, and outcomes were collected from the medical records.
Autoimmune findings were obtained from the same medical records to verify autoimmunity. Autoinflammation was diagnosed after infectious etiology was excluded in patients with inflammation. After this, autoimmune manifestations were grouped according to organ systems. Autoimmune neutropenia, autoimmune cytopenia, autoimmune hemolytic anemia (AHA), and immune thrombocytopenic purpura (ITP) were included in hematologic manifestations. Addison’s disease, hypoparathyroidism, autoimmune thyroiditis, and type 1 diabetes mellitus (DM) were included in endocrinologic manifestations. Chilblain vasculitis, psoriasis, dermatitis, alopecia, and vitiligo were included in dermatologic manifestations. Systemic lupus erythematosus (SLE), juvenile rheumatoid arthritis, scleroderma, and dermatomyositis were included in rheumatologic manifestations. Celiac disease, Crohn’s disease, autoimmune hepatitis, inflammatory bowel disease, and autoimmune enteropathy were included in gastrointestinal manifestations; myasthenia gravis was included in neurologic manifestations. Autoimmune manifestations appearing after allogeneic hematopoietic stem cell transplantation were not included in the study analysis.
This retrospective study was approved by the medical ethics committee of Çukurova University Medical School Hospital on November 27, 2017 (decision No. 2017-167).
Statistical Analysis
All analyses were performed using SPSS 20.0 statistical software package (IBM SPSS Statistics). Categorical variables were expressed as numbers and percentages whereas continuous variables were summarized as mean values and standard deviations, and as median and minimum–-maximum where appropriate. The statistical level of significance for all tests was considered as 0.05.
Results
Patients and clinical findings
The study group comprised 433 patients with PIDs, and 169 (39%) of them were females. The mean age of the patients was 3.2±0.8 years (range: 0–20 years). Median age at onset of symptoms was found as 12 months; 69 (16%) patients died during the study period. Diagnosis of PID was verified via immunologic testing in all patients and genetic testing in 71.1% of the patients. The patients’ epidemiologic data are shown in Table 1.
Table 1 Demographic data of patients with PIDs.
| No. (%) | |
|---|---|
| Number of patients, n (%) | 433 (100) |
| Gender, n (%) Male Female |
263 (60.7) 170 (39.3) |
| Mean age at PID onset (month) | 21.47±28.23 med: 12 |
| Deceased patients, n (%) | 69 (16) |
| Patients with genetic diagnosis, n (%) | 308 (71.1) |
| PID with AI manifestation, n (%) Combined deficiencies CID with associated or syndromic features Predominantly antibody deficiencies Immune dysregulation Defects of phagocytes Defects of innate immunity Autoinflammatory syndromes |
68 (15.7) 101 (23.3) 143 (33) 30 (6.9) 45 (10.4) 24 (5.5) 22 (5.1) |
| Types of AI manifestations Endocrine disorders Hematologic disorders Rheumatologic disorders Skin disorders Gastrointestinal disorders Neurologic disorders |
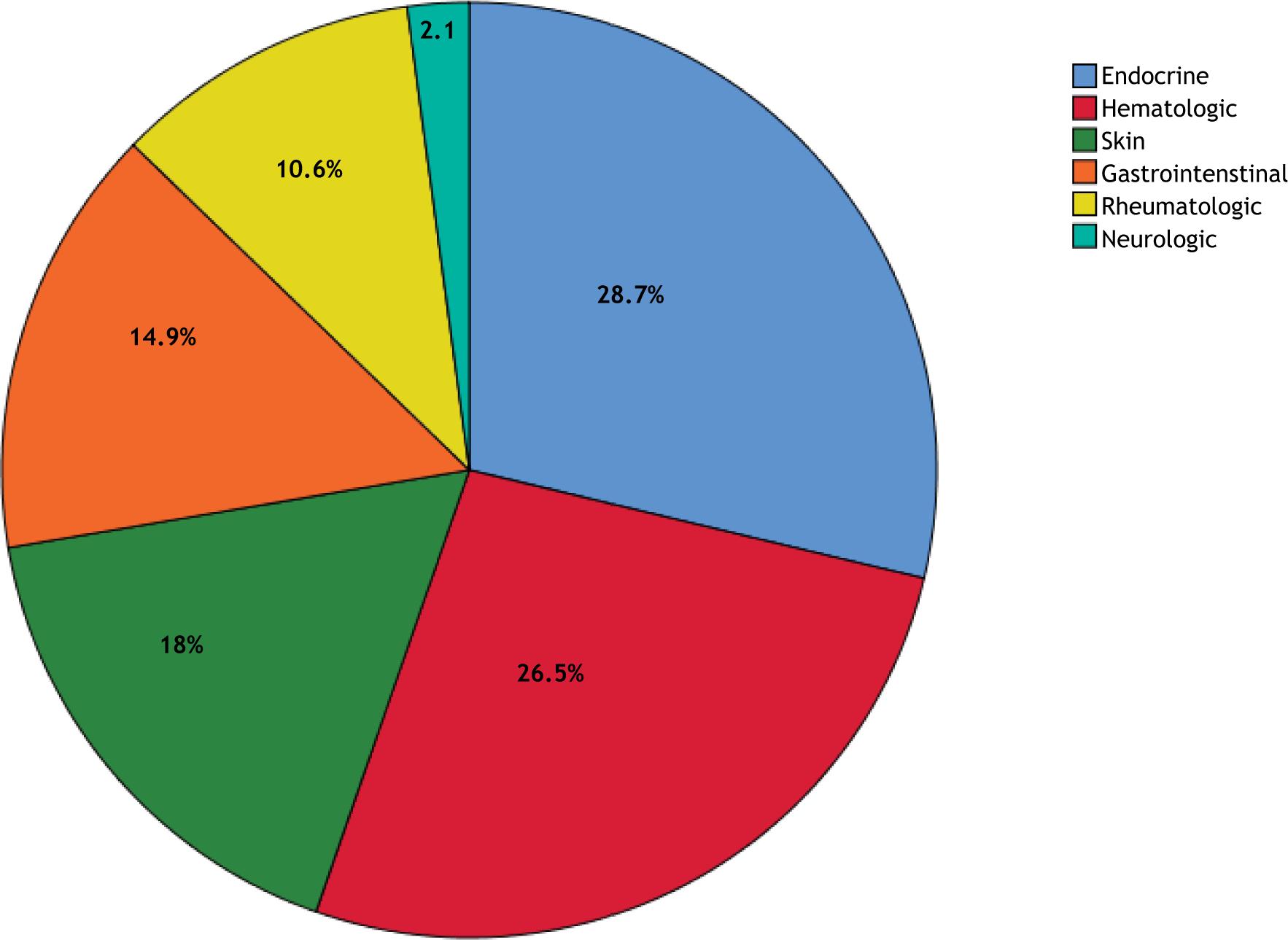
27 (28.7) 25 (26.5) 10 (10.6) 17 (18) 14 (14.9) 2 (2.1) |
PID: primary immunodeficiency disease; AI: autoimmune disease; CID: combined immunodeficiency.
In the autoimmune disease group, the age of onset of immunodeficiency symptoms was 36.50±38.60 months (Median age: 24 months). In the without autoimmune disease findings group, the age was found to be 18.87±25.17 months (median age: 12 months). There was a statistically significant difference in the age of onset of symptoms between the two groups (P = 0.001) (data not shown).
Autoimmune disorders
In all, 69 (15.9%) patients presented with 94 autoimmune manifestations (Table 2). Altogether, 45% of these manifestations were among those presenting symptoms before PID diagnosis, while 55% were documented after establishing the diagnosis. On the whole, 48 patients (69.5%) developed 1 autoimmune manifestation, 17 patients (24.5%) developed 2 such manifestations, and 4 (6%) patients had ≥3 autoimmune manifestations.
Table 2 Characteristics of 69 PIDs with autoimmune disorders.
| PID | Mutation/deletion | Type of autoimmunity | PID | Mutation/deletion | Type of autoimmunity |
|---|---|---|---|---|---|
| IgAD | - | Celiac disease | CVID | - | Type 1 diabetes mellitus |
| IgAD | - | Type 1 diabetes mellitus | CVID | - | SLE |
| IgAD | - | Autoimmune hepatitis | CVID | Cytopenia | |
| IgAD | - | Dermatomyositis | CVID | - | Type 1 diabetes mellitus, alopecia, thyroiditis |
| IgAD | - | Type 1 diabetes mellitus | CVID | - | Cytopenia |
| IgAD | - | Vitiligo | CID | TFRC | Cytopenia |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CID | MALT1 | Dermatitis, Addison’s disease |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | SCID | RAG1 | Cytopenia |
| APECED | AIRE | Hypoparathyroidism | SCID | DCLRE1C | AIHA |
| APECED | AIRE | Hypoparathyroidism | SCID | DCLRE1C | Cytopenia |
| APECED | AIRE | Alopecia, Addison’s disease, enteropathy | SCID | NHEJ1 | AIHA, enteropathy |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CID | WAS | AIHA |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CID | SPINK5 | Skin vasculitis |
| APECED | AIRE | Thyroiditis | Bruton | Btk | JIA |
| APECED | AIRE | Addison’s disease | Bruton | Btk | Skleroderma, autoimmune hepatitis |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CVID | TNFRSF13B | Dermatitis |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CVID | TNFRSF13C | Skin vasculitis |
| APECED | AIRE | Hypoparathyroidism, Addison’s disease | CVID | PIK3CD | Crohn’s disease, cytopenia |
| AT | ATM | JIA | IPEX | FOXP3 | Type 1 diabetes mellitus |
| AT | ATM | ITP | LRBA def. | LRBA | AIHA, ITP, enteropathy |
| AT | ATM | ITP | LRBA def. | LRBA | Enteropathy |
| AT | ATM | AIHA | LRBA def. | LRBA | AIHA, ITP, SLE |
| AT | ATM | JIA | LRBA def. | LRBA | AIHA |
| AT | ATM | ITP | CTLA4 def. | CTLA4 | Enteropathy |
| AT | ATM | Vitiligo | CTLA4 def. | CTLA4 | ITP |
| AT | ATM | Vitiligo | XLP2 | XIAP | Enteropathy |
| AT | ATM | Vitiligo | CMC | STAT1 | AIHA, enteropathy |
| AT | ATM | JIA | CMC | STAT1 | Enteropathy |
| AT | ATM | ITP | CVID | TTC37 | AIHA |
| ALPS | - | ITP, vitiligo | Aicardi | TREX1 | Chilblain lupus |
| ALPS | TNFRSF6 | Chron, psoriasis | Aicardi | TREX1 | Chilblain lupus, central vasculitis |
| CGD | CYBB | JIA | Griscelli | RAB27A | Myastenia gravis, AIHA |
| CVID | - | Cytopenia, enteropathy | HIDS | JIA | |
| MSMD | IL12RB1 | Skin vasculitis | MSMD | IL12RB1 | Skin vasculitis |
| MSMD | IL12RB1 | Skin vasculitis |
IgAD: immunoglobulin A deficiency; APECED: autoimmune polyendocrinopathy, candidiasis, ectodermal dysplasia; AT: ataxia telangiectasia; AIRE: autoimmune regulator; JIA: juvenile idiopatic arthritis; ALPS: autoimmune lymphoproliferative syndrome; AIHA: autoimmune hemolytic anemia; LRBA def.: lipopolysaccharide responsive beige-like anchor deficiency; MSMD: Mendelian susceptibility to mycobacterial disease; CVID: common variable immunodeficiency; MALT1: mucosa-associated lymphoid tissue lymphoma translocation 1 deficiency; ITP: idiopathic thrombocytopenic purpura; CTLA4 def.: cytotoxic T-lymphocyte-associated protein 4 deficiency; IPEX: immunodysregulation polyendocrinopathy enteropathy X-linked; WAS: Wiskott–Aldrich syndrome; CGD: chronic granulomatous disease; CMC: chronic mucocutaneous candidiasis; CID: combined immunodeficiency; SCID: Severe combined immunodeficiency; HIDS: hyperimmunoglobulin D syndrome; SLE: systemic lupus erythematosus.
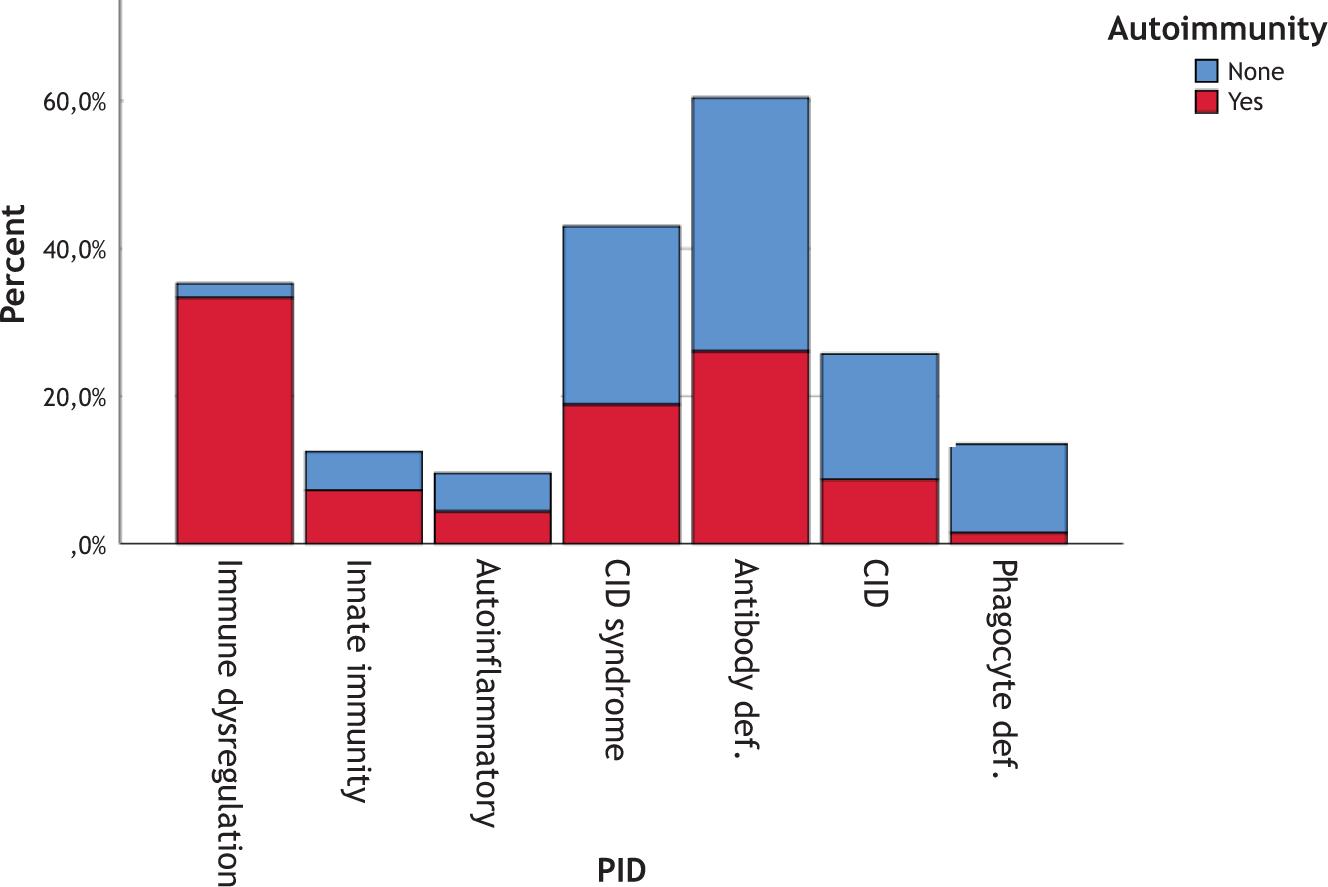
Autoimmune diseases were most prevalent in the subgroup of immune dysregulation of PIDs. They were identified in 23/30 patients with immune dysregulation, 5/24 patients with defects of innate immunity, 13/101 patients with combined immunodeficiency (CID) with associated or syndromic features, 18/143 patients with predominantly antibody deficiencies, 6/68 patients with immunodeficiencies affecting cellular and humoral immunity, 3/22 autoinflammatory disorders, and 1/45 patients with congenital defects of phagocyte number or function (Figure 1).
Figure 1 Distribution of autoimmune diseases according to PID types.
CID: combined immunodeficiency.
In an analysis of AI presentations in patients with PID, 23 different AI diseases appeared: Addison’s disease (n = 10), hypoparathyroidism (n = 10), autoimmune hemolytic anemia (n = 10), autoimmune enteropathy (n = 9), ITP (n = 8), autoimmune cytopenia (n = 7), juvenile idiopatic arthritis (JIA; n = 6), vitiligo (n = 5), type 1 DM (n = 5), skin vasculitis (n = 4), dermatitis (n = 2), Crohn’s disease (n = 2), alopecia (n = 2), autoimmune hepatitis (n = 2), autoimmune thyroiditis (n = 2), chilblain vasculitis (n = 2), SLE (n = 2), psoriasis (n = 1), myasthenia gravis (n = 1), celiac disease (n = 1), dermatomyositis (n = 1), scleroderma (n = 1), and central vasculitis (n = 1); 21 patients had more than one autoimmune disease. Addison’s disease, hypoparathyroidism, and autoimmune hemolytic anemia were common presentations of autoimmunity (endocrine and hematologic, respectively; Figure 2). Autoimmune diseases that developed in patients with PIDs are shown in Table 2. Addison’s disease and/or hypoparathyroidism exist in 12/13 (92.3%) patients with autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy (APECED).
Figure 2 Distribution of autoimmune diseases according to organ systems.
Parenteral immunoglobulin in 46 (66.6%) patients, glucocorticoids in 37 (53.6%), abatacept in 6 (8.7%), mesalazine in 4 (5.8%), methotrexate in 3 (4.3%), tofacitinib in 1 (1.4%), ruxolitinib in 1 (1.4%), mikofenolate mofetil in 1 (1.4%), rapamycin in 1 (1.4%), and canakinumab in 1 (1.4%) patient were prescribed for treatment of autoimmune and autoinflammatory manifestations. Celiac diet was started in one patient, seven patients underwent hematopoietic stem cell transplantation.
Autoinflammatory disorders
Autoinflammation was identified in 22 of the 433 (5.1%) patients with PID, including hyperimmunoglobulin D syndrome (HIDS) (n = 9), Aicardi–Goutieres syndrome (AGS) 1 (n = 6), adenosine deaminase 2 (ADA2) deficiency (n = 3), Blau syndrome (n = 2), tumor necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS) (n = 1), and auto-inflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation (APLAID) (n = 1). Diagnosis of autoinflammatory disorders in all patients was confirmed by genetic testing. Two patients with AGS 1, one of whom having cerebral vasculitis, developed chilblain lupus.
Discussion
We retrospectively analyzed autoimmune and autoinflammatory manifestations in 433 patients with 60 different PIDs enrolled in Southeast Turkey. In this study, 15.9% of the patients with PID had autoimmune manifestations. Addison’s disease, hypoparathyroidism, and autoimmune hemolytic anemia were the most commonly identified autoimmune disorders. Autoinflammatory disorders were present in 5.1% of the patients with PID in our cohort.
In the literature, autoimmune manifestations associated with PID are reported at quite different rates. Patıroğlu et al. showed that among 538 children with PID, autoimmune manifestations were found in 4.6% of patients.7 In another pediatric study, the frequency of autoimmunity was reported to be similar to that in our study regarding patients with PID. Kaplan et al. reported that autoimmune manifestations were determined in 83 patients (10.1%) of the 1036 patients with primary immunodeficiency.8 While the frequency of autoimmunity was 22% in adults and children with PID registered in the Slovenian national PID registry, this ratio was 26.2% in the French national database.9,10 In two centers of North America, where 287 adults and 115 children with PID were evaluated, the frequency of autoimmunity was 27.5% in the adult group and 8.7% in the pediatric group.11 These results obtained from retrospective studies may lead to different rates of autoimmune disease incidences in patients with PID. In addition, the increasing frequency of autoimmunity, especially in adults, requires continuous follow up of patients in this respect.
According to PID classifications, autoimmune signs could appear at different frequencies. Although it is reported that the association of immunodeficiency and autoimmunity is mostly seen in patients with humoral immunodeficiency, different results are reported in the literature.8 In the study conducted by Blazina et al., autoimmune diseases were most common in PIDs, with 100% classified as immune dysregulation diseases, consistent with the results of our study.9 The association of immune dysregulation and autoimmune manifestations in many PIDs has been reported in the literature.12 This overlap is also found in some well-established disorders. It is reported that APECED has been associated with several autoimmune endocrinopathies, including insulin-dependent diabetes mellitus, autoimmune thyroiditis, and ovarian and testicular failure. In addition, these manifestations include adrenal deficiency and parathyroidism, which affect almost all patients with APECED. Autoantibodies against Interleukin (IL)-17, IL-22, and IFNγ have also been shown in the majority of patients with APECED.12 In our study, we detected autoimmune manifestations in almost all patients with APECED, which was in accordance with the literature. In addition, autoimmune findings were highest in PID patients with immune dysregulation.
The chronic infection that develops causes the continuous activation of innate immune response. It has been demonstrated in recent years that the innate immune system plays an important role in autoimmunity.13 Innate immunodeficiencies, which are generally predisposed to infections, can also present with a variable degree of autoimmune manifestations, predominantly autoimmune cytopenias.14 Autoimmune thrombocytopenia, cytopenia, and/or hemolytic anemia are reported as common hematologic findings in patients with PIDs.15 In our study, autoimmune hematologic findings were present in 21 patients (30.4%) in accordance with the literature. These results indicate that patients with cytopenia should be screened for PID. Immunocompromised patients may also develop diseases such as vitiligo, alopecia, SLE, dermatomyositis/polymyositis, arthritis, autoimmune thyroid disease, autoimmune hepatitis, and inflammatory bowel disease.16
Sometimes autoimmune findings could be the first sign of immunodeficiency. As with complement deficiencies or selective IgA deficiency, patients may present with an autoimmune finding without developing immune deficiency symptoms.17 Kaplan et al. reported that autoimmune manifestations occurred in 80.7% of patients before PID diagnosis, and in 19.3% of patients during follow-up.8 In the study conducted by Blazina et al., 81% of patients with PID, who developed autoimmunity, presented with autoimmune manifestations.9 In the two studies evaluating patients with common variable immunodeficiency (CVID) before the diagnosis of immunodeficiency, hematologic diseases were the first findings in 54% and 62% of patients, respectively.18,19 Multiple autoimmune disease findings may also develop in patients with PID. In the literature, findings of autoimmune disease in more than one system are reported in approximately one-third of patients with autoimmunity with PID.8 In our study, 30.5% of the patients had multiple autoimmune diseases, which was in accordance with the literature. Moreover, in this study, 45% of the patients with autoimmune diseases had autoimmune disease findings before diagnosis of PID. In addition, the age of onset of immunodeficiency symptoms was higher in patients with autoimmunity than those without it. This situation suggests the necessity of PID research, especially in patients with early onset and/or multiple autoimmune manifestations.
Autoinflammatory diseases are caused by primary disorder of innate immune system. These disorders are characterized by recurrent attacks of fever, polyserositis, arthritis, and cutaneous symptoms. While there are differences between autoinflammatory and autoimmune diseases, they also share many similar clinical manifestations. These clinical manifestations sometimes overlap, thus obscuring the diagnosis.20 The chilblain lesions seen in about half of AGS patients provide a useful clinical manifestation of the disease and show an immune pathology. Rice et al. described heterozygous TREX1 mutations in an autosomal dominant cutaneous form of SLE called “familial chilblain lupus”.21 The exact functions of the TREX1 complex are unknown, but it is thought to play a role in removing nucleic acid species produced during apoptosis. A defect in this process results in the activation of innate immune system.21 In the present study, both autoinflammatory and autoimmune findings were present in two (33.3%) patients with AGS. Therefore, in the presence of autoimmune manifestations accompanied by periodic findings, the presence of autoinflammatory diseases should be investigated.
The limitations of this study include its retrospective nature and inability to perform long-term follow-up of PID patients due to early death. It contributes to the literature because it includes rarely seen autoinflammatory diseases in the pediatric age group.
Conclusions
In conclusion, autoimmune and autoinflammatory manifestations are relatively common findings in patients with PID. Our results emphasize a close relationship between autoimmunity and PID. Autoimmunity might be a clue to underlying immune dysregulation during the evaluation of a patient with severe and recurrent infections.
Conflict of interest
The authors have no ethical and financial conflicts to disclose.
REFERENCES
1. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: Two sides of the same coin? Nat Rev Rheumatol. 2017 Dec 19;14(1):7–18. 10.1038/nrrheum.2017.198
2. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, Members of the CEREDIH French PID Study Group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2017 Nov;140(5):1388–93.
3. Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Ann Allergy Asthma Immunol. 2019 Nov;123(5):454–60. 10.1016/j.anai.2019.07.014
4. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun. 2019 May;99:52–72. 10.1016/j.jaut.2019.01.011
5. Bousfih A, Jeddane L, Picard C, Ailal F, Gaspar HB, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2018 Jan 38(1):129–43. 10.1007/s10875-017-0465-8
6. Műzes G, Sipos F. Primary immunodeficiency and autoimmune diseases. Orv Hetil. 2018 Jun;159(23):908–18. 10.1556/650.2018.31064
7. Patıroğlu T, Gungor HE, Unal E. Autoimmune diseases detected in children with primary immunodeficiency diseases: Resultsfrom a reference centre at middle Anatolia. Acta Microbiol Immunol Hung. 2012;59:343–53. 10.1556/amicr.59.2012.3.5
8. Kaplan MY, Ozen S, Akcal O, Gulez N, Genel F. Autoimmune and inflammatory manifestations in pediatric patients with primary immunodeficiencies and their importance as a warning sign. Allergol Immunopathol. 2020;48(6):701–10. 10.1016/j.aller.2020.02.009
9. Blazina S, Markelj G, Jeverica AK, Toplak N, Bratanič N, Jazbec J, et al. Autoimmune and inflammatory manifestations in 247 patients with primary immunodeficiency—A report from the Slovenian national registry. J ClinImmunol. 2016;36:764–73. 10.1007/s10875-016-0330-1
10. Azizi G, Tavakolinia M, Rafiemanesh H, Kiaee F, Yazdani R, Heydari A, et al. Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Rev Clin Immunol. 2017;13:1099–1106. 10.1080/1744666X.2017.1384312
11. Pellegrino M, Bellacchio E, Dhamo R, Frasca F, Betterle C, Fierabracci A. A novel homozygous mutation of the AIRE Gene in an APECED Patient from Pakistan: Case report and review of the literature. Front Immunol. 2018 Aug 13;9:1835-41. 10.3389/fimmu.2018.01835
12. Humbert L, Cornu M, Proust-Lemoine E, Bayry J, Wemeau JL, Vantyghem MC, et al. Chronic mucocutaneous candidiasis in autoimmune Polyendocrine Syndrome Type 1. Front Immunol. 2018 Nov 19;9:2570-8. 10.3389/fimmu.2018.02570
13. Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016 Feb;46(2):269–80. 10.1002/eji.201545839
14. Croker BA, Silke J, Gerlic M. Fight or flight—Regulation of emergency hematopoiesis by pyroptosis and necroptosis. Curr Opin Hematol. 2015 Jul;22(4):293–301. 10.1097/MOH.0000000000000148
15. Azizi G, Abolhassani H, Kiaee F, Tavakolinia M, Rafiemanesh H, Yazdani R, et al. Autoimmunity and its association with regulatory T cells and B cell subsets in patients with common variable immunodeficiency. Allergol Immunopathol. 2018;46(2):127–35. 10.1016/j.aller.2017.04.004
16. Singh K, Chang C, Gershwin ME. IgA deficiency and autoimmunity. Autoimmun Rev. 2014 Feb;13(2):163–77. 10.1016/j.autrev.2013.10.005
17. Conigliaro P, Triggianese P, Ballanti E, Perricone C, Perricone R, Chimenti MS. Complement, infection, and autoimmunity. Curr Opin Rheumatol. 2019 Sep;31(5):532–41. 10.1097/BOR.0000000000000633
18. Michel M, Chanet V, Galicier L, Ruivard M, Levy Y, Hermine O, et al. Autoimmune thrombocytopenic purpura and common variable immunodeficiency: Analysis of 21 cases and review of the literature. Medicine. 2004;83:254–63. 10.1097/01.md.0000133624.65946.40
19. Wang J, Cunningham-Rundles C. Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). J Autoimmun. 2005;25:57–62. 10.1016/j.jaut.2005.04.006
20. Georgin-Lavialle S, Fayand A, Rodrigues F, Bachmeyer C, Savey L, Grateau G. Autoinflammatory diseases: State of the art. Presse Med. 2019;48:25–48. 10.1016/j.lpm.2018.12.003
21. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi–Goutieres syndrome. Am J Hum Genet. 2007;80:811–15. 10.1086/513443