
Download
ORIGINAL ARTICLE
Pulmonary manifestations in a cohort of patients with inborn errors of immunity: an 8-year follow-up study
Mahshid Movahedia, Mahnaz Jameeb, c, Hosseinali Ghaffaripoura, Farzad Nooria, Mehdi Ghainid, Shabnam Eskandarzadehd, Javad Enayata, Golnaz Eslamiand, Guitti Pourdowlate, Niusha Sharifinejadf, Mihan Poorabdollaha, Seyed Alireza Nadjia, Mazdak Fallahia, Zahra Daneshmandia, Jalal Heshmatniae, Alireza Eslaminejade, Atefeh Fakhariane, Maryam Vasheghanie, Afshin Monirig, Maryam Sadat Mirenayate, Payam Tabarsig, Majid Marjanig, Nima Rezaeih, i, j, Mikko R. J. Seppänenk, Davood Mansouria, g, Seyed Alireza Mahdaviania*, Ali Akbar Velayatia
aPediatric Respiratory Diseases Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
bPediatric Infections Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran
cPediatric Nephrology Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran
dDepartment of Allergy and Clinical Immunology, Mofid Children’s Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran
eChronic Respiratory Diseases Research Center, National Research Institute of Tuberculosis and Lung Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran
fStudent Research Committee, Alborz University of Medical Sciences, Karaj, Iran
gClinical Tuberculosis and Epidemiology Research Centre, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
hResearch Center for Immunodeficiencies, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran
iDepartment of Immunology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
jPrimary Immunodeficiency Diseases Network (PIDNet), Universal Scientific Education and Research Network (USERN), Tehran, Iran
kRare Disease and Pediatric Research Centers, Hospital for Children and Adolescents and Adult Immunodeficiency Unit, Inflammation Center, University of Helsinki and HUS Helsinki University Hospital, Helsinki, Finland
Abstract
Background Inborn errors of immunity (IEIs) are a group of congenital diseases caused by genetic defects in the development and function of the immune system. The involvement of the respiratory tract is one of the most common presentations in IEIs.
Methods Overall, 117 patients with diagnosed IEIs were followed-up within 8 years at the National Research Institute of Tuberculosis and Lung Diseases (NRITLD). Demographic, clinical, and laboratory data were collected in a questionnaire. Pulmonary function test (PFT), chest X-ray (CXR), and high-resolution computed tomography (HRCT) scans were obtained where applicable.
Results Our study population consisted of 48 (41%) patients with predominantly antibody deficiencies (PADs), 39 (32%) patients with congenital defects of phagocytes, 14 (11.9%) patients with combined immunodeficiency (CID), and 16 (14%) patients with Mendelian susceptibility to mycobacterial diseases (MSMD). . Recurrent pneumonia was the most common manifestation, while productive cough appeared to be the most common symptom in almost all diseases. PFT showed an obstructive pattern in patients with PAD, a restrictive pattern in patients with CID, and a mixed pattern in patients with CGD. HRCT findings were consistent with bronchiectasis in most PAD patients, whereas consolidation and mediastinal lesions were more common in the other groups.
Conclusions Pulmonary manifestations vary among different groups of IEIs. The screening for lung complications should be performed regularly to reveal respiratory pathologies in early stages and follow-up on already existing abnormalities.
Key words: inborn errors of immunity, primary immunodeficiency, respiratory tract infections, pulmonary, CT, PFT
*Corresponding author: Seyed Alireza Mahdaviani, Pediatric Respiratory Diseases Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran. Email address: [email protected]
Received 18 March 2021; Accepted 15 August 2021; Available online 1 January 2022
Copyright: Movahedi M, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Inborn errors of immunity (IEIs) are a group of congenital diseases caused by genetic defects in the development and functioning of the body’s defense mechanisms.1 They display recurrent infections, lymphoproliferation, autoimmunity, autoinflammation, early onset severe atopy, and/or malignancies.2 One of the most prominent organs involved in IEIs is the respiratory tract, causing excess morbidity and mortality in the affected.3 Respiratory tract complications include infectious (e.g., pneumonia, sinusitis, otitis media) and noninfectious (e.g., airway obstruction, interstitial lung disease, bronchiectasis, benign lymphoproliferation, malignancies) manifestations.4 A better understanding of pulmonary complications in IEIs will be helpful for the timely diagnosis of IEIs and may help prevent their further progression.5 In previous studies, several patterns of respiratory tract involvement in a variety of IEIs have been suggested.6 However, comparative studies of clinical and radiological manifestations among different types of IEIs are few. In order to better evaluate and describe pulmonary manifestations of patients with IEIs, we compared the pulmonary characteristics in a cohort of children suffering from variable IEIs, followed at our institution.
Methods and Materials
We retrospectively reviewed the medical records of patients diagnosed with IEIs from 2010 to 2018, who were referred to the National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Masih Daneshvari Hospital, Tehran, Iran. Patients with predominantly antibody deficiencies (PADs) including common variable immunodeficiency (CVID), hyper IgM syndrome (HIGM), X-linked agammaglobulinemia (XLA), as well as congenital defects of phagocytes, combined immunodeficiency (CID), and Mendelian susceptibility to mycobacterial diseases (MSMD) who were diagnosed and treated at this center enrolled in this study. When no genetic diagnosis was available, the underlying IEIs were diagnosed according to the criteria of the European Society for Immunodeficiencies (ESID) Registry Working Party Written informed consent was obtained from all patients (and/or their guardians). The study was accepted by the institutional review/ethics board. Patients or their guardians completed a questionnaire for demographic information including age, sex, age at onset of symptoms, age at diagnosis, presence of familial history, and consanguinity. The clinical manifestations, complications, and comorbidities were extracted from the medical records and thus represented physician-reported morbidities. Diagnostic delay was defined as the time between the onset of symptoms and the time of diagnosis. The patients’ blood samples were tested quantitatively for lymphocyte subset and immunoglobulin levels, and compared with age-matched normal reference values. Isohemagglutinin titer, anti-diphtheria, and anti-tetanus were evaluated.
Pulmonary function tests (PFTs) were performed according to the American Thoracic Society recommendations7 in patients who were older than 6 years and cooperative, with patients tested in the seated position within a volume displacement body plethysmograph. Respiratory parameters including FEV1, FVC, FEV1/FVC, and maximal mid-expiratory flow (MMEF 25–75%) were recorded for each patient. PFT was not performed in MSMD patients due to either age limitation or positive mycobacterium test; therefore, they were excluded in the analysis of PFT patterns. Standard posteroanterior and lateral chest X-ray, as well as high-resolution computed tomography (HRCT) scans, were obtained in all patients. Data were presented as minimum, maximum, and mean (standard deviation) using SPSS software (v. 25.0, Chicago, IL). Epidemiologic, clinical, imaging, and laboratory data were compared among the patient groups.
Results
In total, 117 patients (71 males, 46 females) aged 1–54 years, including 48 (41%) patients with PADs (CVID = 43 (36.7%), HIGM = 3 (2.6%) (including two patients with pathogenic AICDA mutations), XLA = 2 (1.7%)), 39 (32%) patients with congenital defects of phagocytes (CGD [chronic granulomatous disease] = 38 (32.5%), congenital cyclic neutropenia = 1 (0.8%)), 14 (11.9%) patients with CID (severe combined immunodeficiency (SCID) = 5 (4.3%), hyper IgE syndrome (HIES) = 5 (4.3%), ataxia-telangiectasia (AT) = 3 (2.6%), autoimmune poly-endocrinopathy candidiasis ectodermal dystrophy (APECED) = 1 (0.8%)), and 16 (14%) patients with MSMD were enrolled in this study (Table 1).
Table 1 Clinical manifestions in the study population.
| Diagnosis | CVID (n = 43) | HIGM (n = 3) | XLA (n = 2) | CGD (n = 38) | CN (n = 1) | CID (n = 14) | MSMD (n = 16) |
|---|---|---|---|---|---|---|---|
| Signs and symptoms | |||||||
| Dyspnea | 10 | 1 | 1 | 19 | 1 | 8 | 2 |
| Cough | 23 | 2 | 2 | 31 | 1 | 14 | 4 |
| Sputum | 17 | 0 | 2 | 17 | 1 | 10 | 1 |
| Comorbidities | |||||||
| Recurrent pneumonia | 28 | 3 | 2 | 30 | 0 | 10 | 3 |
| Rhinosinusitis | 28 | 2 | 2 | – | 1 | 4 | 0 |
| Otitis media | 16 | 3 | 1 | – | 0 | 2 | 0 |
| Lymphadenopathy | 4 | 3 | 0 | – | 0 | 2 | 16 |
| Lymphoma | 3 | 0 | 0 | – | 0 | 2 | 0 |
| Clubbing | 3 | 0 | 2 | – | 0 | 3 | 0 |
| Oral candidiasis | 0 | 0 | 0 | – | 0 | 11 | 0 |
| Tonsillitis | 0 | 0 | 0 | – | 1 | 9 | 0 |
| Perianal abscess | 0 | 0 | 0 | – | 1 | 0 | 0 |
CVID: common variable immunodeficiency, HIGM: hyper IgM syndrome, XLA: X-linked agammaglobulinemia, CGD: chronic granulomatous disease, CN: congenital neutropenia, CID: combined immunodeficiency, MSMD: Mendelian susceptibility to mycobacterial diseases.
Predominantly antibody deficiency
Among the patients with PADs, there were 27 (56.3%) male and 21 (43.7%) female patients. There was no gender bias within the CVID, HIGM, and XLA groups (P > 0.05). CVID presented with the latest mean age of onset (11.7 years) and diagnosis (25.6 years) and the longest diagnostic delay (13.6 years) among IEIs. No familial history of early demise was reported.
The most common clinical symptoms in all PAD patients were cough (n = 27, 56.3%) and sputum (n = 19, 39.6%). Respiratory tract infections included recurrent pneumonia (n = 33, 68.8%), rhinosinusitis (n = 32, 69.6%), and otitis media (n = 20, 43.5%). Other less frequent manifestations were lymphadenopathy (n = 7, 14.2%), lymphoma (n = 3, 6.1%), clubbing (n = 5, 10.2%), and opportunistic infections (n = 1, 2.0%).
Bronchiectasis (n = 28, 58.3%) was the most common HRCT scan finding (Figure 1A), followed by bronchial wall thickening (n = 11, 22.9%) and consolidation (n = 9, 18.8%). Bronchiectasis complicated 25 (58.1%) CVID, one HIGM, both XLA patients, and was bilateral in 64.3% (18 of 28) of them. The most frequent site of bronchiectasis in all patients with PAD was the right middle lobe. Overall, PFTs exhibited an obstructive pattern in 10 (31.3%), restrictive pattern in 8 (25%), and mixed pattern in 2 (6.2%) patients. Ten out of 29 CVID patients (34.5%) with available PFT displayed a purely obstructive pattern , while five (17.2%) and two (7.4%) CVID patients had restrictive and mixed pattern, respectively. All patients with HIGM and XLA had a restrictive pattern.
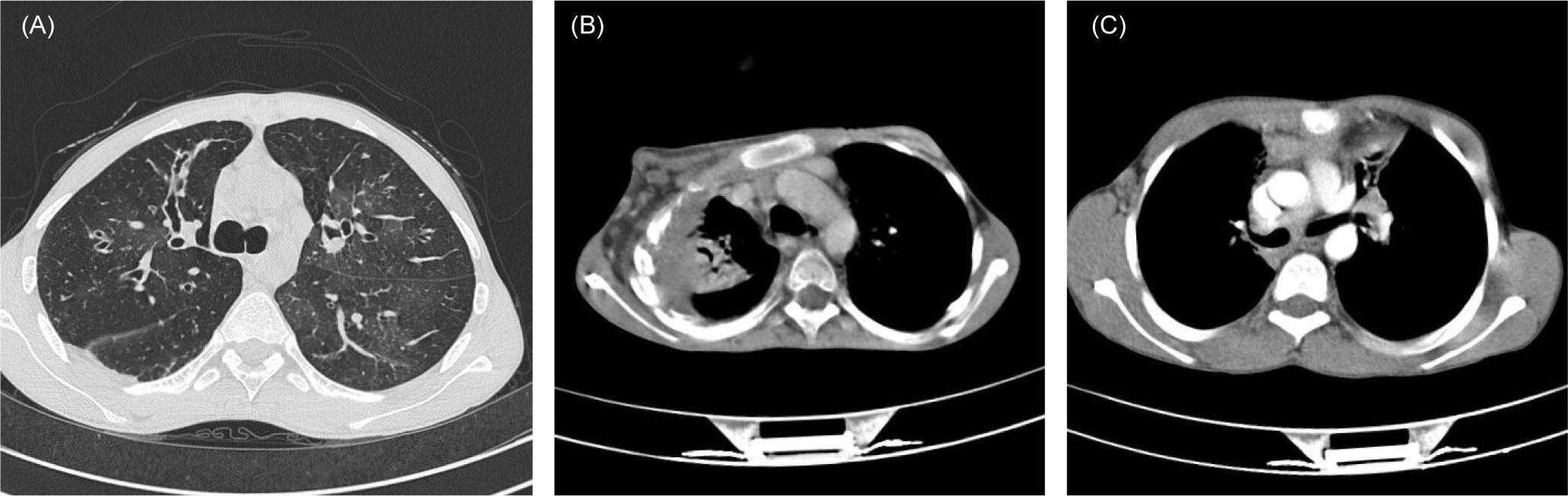
Figure 1 Axial chest HRCT images depicting the most common findings in patients with inborn errors of immunity. (A) Tubular and varicoid bronchiectasis and patchy ground-glass opacities in bilateral middle and lower lung lobes of a patient with X-linked agammaglobulinemia (XLA). (B) Focal collapse consolidation with invasion to the chest wall and associated osteolytic bone lesions in a patient with CGD. (C) Bilateral mediastinal lymphadenopathy in a patient with MSMD.
Combined immunodeficiency
Six (42.9%) male and eight (57.1%) female patients were diagnosed with CID. Six (42.9%) patients were born to consanguineous parents. The mean age of diagnosis and diagnostic delay were 6.7 and 6.3 years, respectively. Cough (n = 14, 100%), sputum (n = 10, 71.4%), and dyspnea (n = 8, 57.1%) were among the common presentations. Oral candidiasis was reported to be the most common comorbidity in CID patients (n = 11, 78.6%), followed by recurrent pneumonia (n = 10, 71.4%), tonsillitis (n = 9, 64.3%), rhinosinusitis (n = 4, 28.6%), clubbing (n = 3, 21.4%), otitis media (n = 2, 14.3%), lymphadenopathy (n = 2, 14.3%), and lymphoma (n = 2, 14.3%). HRCT scans were mostly compatible with consolidation (n = 6, 42.9%).
The most frequent sites of bronchiectasis in CID patients were the lower lobes and middle lobe (in two patients each). In PFT, the restrictive pattern (two of five, 40%) was the commonest.
Congenital defects of phagocytes
CGD was diagnosed by nitro blue tetrazolium (NBT) assay and included 30 (79%) male and 8 (21%) female patients, with a mean age of onset of 6.7 years and diagnosis of 12.8 years. Dihydrorhodamine (DHR) test was performed in all patients and was abnormal in 11 out of 27 (28.9%) patients. Twenty-four (63.1%) CGD patients were born to consanguineous parents. Familial history of early demise was reported in 14 (34.2%) patients. Thirty (78.9%) CGD patients were diagnosed with pneumonia, mostly at first presentation. Other involved organs included the lymphatic system, skin, and gastrointestinal system.
Consolidation (n = 17, 44.7%), bronchiectasis (n = 11, 28.9%), and nodular infiltration (n = 11, 28.9%) were the most reported finding in CGD patients, especially in the right upper lobe of the lung (Figure 1B). A mixed restrictive–obstructive pattern in PFT was the most common (n = 8, 21%). The patient with congenital cyclic neutropenia suffered from productive cough, dyspnea, sinusitis, and perianal abscess, and presented with nodular infiltration in the HRCT and obstructive pattern in the PFT.
Defects in Intrinsic and Innate immunity
Eight (50%) male and eight (50%) female patients with MSMD were found. Among the diagnostic groups, MSMD patients had the earliest age of onset (0.5 year) and diagnosis (4.5 years), with a mean diagnostic delay of 4.7 years. Thirteen (81.2%) patients were born to consanguineous families. Patients with MSMD presented with cough (n = 4, 25%), dyspnea (n = 2, 12.5%), and sputum (n = 1, 6.2%), and pulmonary manifestation was limited to recurrent pneumonia (n = 3,18.8%) with no signs of sinusitis or otitis. Lymph node involvement was observed in all patients in axillary (75%), mediastinal (12.5%), upper abdominal (31.2%), and cervical (12.5%) regions. Consolidation (n = 3, 18.8%) and mediastinal lesions (n = 8, 18.8%) were the most reported HRCT scan findings (Figure 1C).
Discussion
Herein, we investigated pulmonary manifestations among different groups of IEI. Most of the patients suffered from some lung involvement, often early in the course of the disease and with considerable impact on morbidity. Altogether, recurrent respiratory infections seemed frequently responsible for chronic symptoms, frequent use of antibiotics, hospitalizations, impaired quality of life, and anatomical lung abnormalities, resulting in vicious cycles. Susceptibility to lung diseases varied significantly among our patients, despite the relative lack of previous formal comparative studies and suggesting further ones in large and well-defined cohorts.8
In our cohort, recurrent pneumonia along with sinusitis was the most common manifestation. The most frequent clinical symptom in patients with PAD, CID, CGD, and MSMD was productive cough. In one previous survey, the most prevalent respiratory symptom in patients with XLA and CVID was also persistent cough (30.0%), shown to be significantly associated with the need to continue antibiotic treatment compared to those without cough.9 In addition, previous studies have shown that respiratory tract infections are the most common infectious complication among CGD and CID patients, detected in about 80 and 69% of them, respectively.10,11
Previous studies have also shown that among humoral immunodeficiency patients with HRCT abnormalities, bronchiectasis and bronchial wall thickening are the most common findings and reported in 14–60%,12 seen most frequently in the middle or lower lobe.9,12 Likewise, we found middle lobe bronchiectasis (n = 28, 58.3%) to be the most common HRCT finding. However, in CGD patients, consolidation, bronchiectasis, and nodular infiltration, most commonly in the right upper lobe, were the most frequently reported findings, corroborating earlier studies.13 In the acute phase of infections, consolidations mainly involve the lower lobes, which gradually increase in size and tend to transfer from the left lower lobe to the left upper lobe.14
In PFT, observed impairment interestingly was associated with IEI classification; an obstructive pattern in patients with PAD, restrictive pattern in patients with CID, and mixed pattern in patients with CGD predominated. As patients with IEIs have a high risk of multiple pulmonary complications that may present with similar symptoms (cough, dyspnea), spirometry is essential in the diagnosis of airway diseases such as asthma or bronchiectasis. Full PFTs, including spirometry, lung volume testing, and diffusing capacity of the lungs for carbon monoxide (DLCO), can noninvasively provide information when restrictive or in IEIs all frequent infiltrative lung diseases were suspected.15
The weaknesses of our study include its retrospective design, the relatively small numbers of patients studied, the unavoidable use of clinical diagnostic criteria for monogenic IEIs where there was no access to exact genetic diagnosis, and the use of physician-reported comorbidities. However, since patients’ respiratory symptoms and findings were common and prominent, our study importantly highlights the necessity to follow-up and screen systematically for respiratory complications. Unfortunately, currently, there are no established consensus guidelines on the routine assessment and prevention of pulmonary complications in these patients.16 The screening for lung complications should be regularly performed to reveal respiratory pathologies at early stages and to monitor the already existing abnormalities in IEI patients. Based on our findings, and to minimize radiation exposure, we recommend HRCT when deemed clinically necessary due to symptoms or PFT findings. Apart from its use in acute lower respiratory tract infections, HRCT currently represents the gold standard for detecting bronchiectasis, interstitial lung diseases (ILDs), and malignancy. However, considering the extra radiation dose imposed on patients who frequently have an increased risk for malignancies, it is recommended to perform HRCT on a clinical basis, as guided by pre-assessment of PFT patterns.17
Compliance with Ethical Standards
The present study was conducted according to the principles stated in the Helsinki Declaration and ethical standards of the Shahid Beheshti University of Medical Sciences committee (IR.SBMU.NRITLD.REC.1396.324).
Funding
The authors received no specific funding for this research.
Conflict of Interest
The authors declare that they have no conflict of interest.
REFERENCES
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. 10.1007/s10875-019-00737-x
2. Tavakol M, Jamee M, Azizi G, Sadri H, Bagheri Y, Zaki-Dizaji M, et al. Diagnostic approach to the patients with suspected primary immunodeficiency. Endocr Metab Immune Disord Drug Targets. 2020;20(2):157–171. 10.2174/1871530319666190828125316
3. Reisi M, Azizi G, Kiaee F, Masiha F, Shirzadi R, Momen T, et al. Evaluation of pulmonary complications in patients with primary immunodeficiency disorders. Eur Ann Allergy Clin Immunol. 2017;49(3):122–128.
4. Yazdani R, Abolhassani H, Asgardoon MH, Shaghaghi M, Modaresi M, Azizi G, et al. Infectious and noninfectious pulmonary complications in patients with primary immunodeficiency disorders. J Investig Allergol Clin Immunol. 2017;27(4):213–224. 10.18176/jiaci.0166
5. Jesenak M, Banovcin P, Jesenakova B, Babusikova E. Pulmonary manifestations of primary immunodeficiency disorders in children. Front Pediatr. 2014;2:77. 10.3389/fped.2014.00077
6. Ramzi N, Jamee M, Bakhtiyari M, Rafiemanesh H, Zainaldain H, Tavakol M, et al. Bronchiectasis in common variable immunodeficiency: a systematic review and meta-analysis. Pediatr Pulmonol. 2020;55(2):292–299. 10.1002/ppul.24599
7. Graham BL, Steenbruggen I, Miller MR, Barjaktarevic IZ, Cooper BG, Hall GL, et al. Standardization of spirometry 2019 update. An Official American Thoracic Society and European Respiratory Society Technical Statement. Am J Respir Crit Care Med. 2019;200(8):e70–e88. 10.1164/rccm.201908-1590ST
8. Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors beyond lack of antibody govern pulmonary complications in primary antibody deficiency. J Clin Immunol. 2019;39(4):440–447. 10.1007/s10875-019-00640-5
9. Costa-Carvalho BT, Wandalsen GF, Pulici G, Aranda CS, Sole D. Pulmonary complications in patients with antibody deficiency. Allergol Immunopathol (Madr). 2011;39(3):128–132. 10.1016/j.aller.2010.12.003
10. Khanna G, Kao SC, Kirby P, Sato Y. Imaging of chronic granulomatous disease in children. Radiographics. 2005;25(5):1183–1195. 10.1148/rg.255055011
11. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. 2018;141(4):1450–1458. 10.1016/j.jaci.2017.06.049
12. Bierry G, Boileau J, Barnig C, Gasser B, Korganow AS, Buy X, et al. Thoracic manifestations of primary humoral immunodeficiency: a comprehensive review. Radiographics. 2009;29(7):1909–1920. 10.1148/rg.297095717
13. Lee M, Lee MS, Lee JS, Ko SY, Jeong SY. Spectrum of imaging findings of chronic granulomatous disease: a single center experience. Diagn Interv Radiol. 2017;23(6):472–477. 10.5152/dir.2017.17264
14. Godoy MC, Vos PM, Cooperberg PL, Lydell CP, Phillips P, Muller NL. Chest radiographic and CT manifestations of chronic granulomatous disease in adults. AJR Am J Roentgenol. 2008;191(5):1570–1575. 10.2214/AJR.07.3482
15. Nonas S. Pulmonary manifestations of primary immunodeficiency disorders. Immunol Allergy Clin North Am. 2015;35(4):753–766. 10.1016/j.iac.2015.07.004
16. Jolles S, Sánchez-Ramón S, Quinti I, Soler-Palacín P, Agostini C, Florkin B, et al. Screening protocols to monitor respiratory status in primary immunodeficiency disease: findings from a European survey and subclinical infection working group. Clin Exp Immunol. 2017;190(2):226–234. 10.1111/cei.13012
17. Cinetto F, Scarpa R, Rattazzi M, Agostini C. The broad spectrum of lung diseases in primary antibody deficiencies. Eur Respir Rev. 2018;27:149. 10.1183/16000617.0019-2018