
Download
ORIGINAL ARTICLE
Knockdown of OLFM4 protects cardiomyocytes from sepsis by inhibiting apoptosis and inflammatory responses
Hailu Chen, Shuna Liu, Guihua Fang*
Department of Infectious Diseases and Tropical Diseases, The Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
Abstract
Sepsis is a systemic inflammatory response that can result in cardiac insufficiency or heart failure known as septic myocardial injury. A previous study identified OLFM4 as an important gene in sepsis through bioinformatics analysis. However, there is limited research on the regulatory functions of OLFM4 in sepsis-triggered myocardial injury, and the related molecular mechanisms remain unclear. In this study, the protein expression of OLFM4 was found to be significantly elevated in LPS-stimulated H9C2 cells, and its suppression enhanced cell proliferation and reduced cell apoptosis in LPS-triggered H9C2 cells. The inflammatory factors TNF-α, IL-6, and IL-1β were increased after LPS treatment, and these effects were mitigated after silencing OLFM4. Moreover, it was confirmed that inhibition of OLFM4 attenuated the NF-κB signaling pathway. In conclusion, the knockdown of OLFM4 protected cardiomyocytes from sepsis by inhibiting apoptosis and inflammatory responses via the NF-κB pathway. These findings provide important insights into the regulatory functions of OLFM4 in the progression of septic myocardial injury.
Key words: myocardial injury, NF-κB pathway, OLFM4, sepsis
*Corresponding author: Guihua Fang, Department of Infectious Diseases and Tropical Diseases, The Affiliated Hospital of Guangdong Medical University, No. 57 Renmin Avenue South, Xiashan, Zhanjiang, Guangdong, China. Email address: [email protected]
Received 1 June 2024; Accepted 4 August 2024; Accepted online 1 September 2024
Copyright: Chen H, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Sepsis, an overwhelming systemic inflammatory response triggered by bacterial infection, is a complication in critically ill patients that can progress to multiple organ dysfunction syndrome (MODS).1 Various treatments have been employed to improve this condition, including blood purification techniques to remove inflammatory mediators and cytokines.2,3 Despite these interventions, the limited treatment options available indicate that sepsis remains a leading cause of high mortality among hospitalized patients globally.4 Cardiac insufficiency is a common occurrence in sepsis patients.5 Studies have shown that the mortality rate for sepsis patients with myocardial injury is approximately 80%, significantly higher than the 20% mortality rate for those without myocardial injury.6 Therefore, understanding the pathogenesis and identifying novel targets for sepsis--induced myocardial injury is significant.
Olfactomedin 4 (OLFM4, also known as GW112 or hGC-1) is a granule protein and a member of the olfactomedin family.7 OLFM4 exhibits dysregulated expression in various inflammatory diseases and is involved in their regulation. For instance, OLFM4 deletion modulates P62-dependent mitophagy, exacerbating nonalcoholic fatty liver disease in mice.8 In addition, OLFM4 can induce dysplasia and inhibit metastasis in colon cancer.9 Quantitative proteomic analysis of synovial tissue has revealed that OLFM4 can exacerbate inflammation in rheumatoid arthritis.10 Moreover, Qinbaohong Zhike oral liquid has been reported to suppress OLFM4, thereby alleviating lipopolysaccharide (LPS)-induced acute lung injury in rats.11 In sepsis-mediated acute respiratory distress syndrome (ARDS)/acute lung injury (ALI), OLFM4 has been shown to affect the NF-κB signaling pathway, thereby improving lung epithelial cell function.12 Notably, a previous study using bioinformatics analysis of three GEO databases identified OLFM4 as a crucial gene in sepsis.13 Therefore, we hypothesized that OLFM4 might also regulate sepsis-induced myocardial injury.
This study investigated the regulatory functions and associated pathways of OLFM4 in sepsis-induced myocardial injury. The findings suggest that OLFM4 may be a potential therapeutic target for ameliorating sepsis-induced myocardial injury.
Materials and Methods
Cell lines and treatments
H9C2 cells were purchased from ATCC (Manassas, VA, USA) and cultured in DMEM containing 10% fetal bovine serum (FBS, Gibco, USA) in an incubator (37°C, 5% CO2, humidified). Lipopolysaccharide (LPS) (5 μg/mL, Sigma-Aldrich, USA) was used to treat H9C2 cells to establish a sepsis--induced myocardial injury model.
Cell transfection
siRNAs targeting OLFM4 (si-OLFM4) and negative control -(si-NC) were purchased from GenePharma (Shanghai, China) and transfected into H9C2 cells using Lipofectamine 2000 (Invitrogen, USA).
Western blotting
Proteins extracted from H9C2 cells were lysed using RIPA buffer. The proteins were separated using 10% SDS-PAGE and then transferred to PVDF membranes (Beyotime, Shanghai, China). After blocking, the membranes were incubated with primary antibodies against OLFM4 (1 µg/mL; ab85046; Abcam, Shanghai, China), p-p65 (1:1000; ab76302), p65 (0.5 µg/mL; ab16502), p-IkBα (1:1000; ab92700), IkBα (1:500; ab76429), p-p65 (1:500; ab28849), p65 (1:1000; ab32360), and GAPDH (1:1000; ab8245) for 12 hours. Subsequently, the appropriate secondary antibodies (1:1000; ab7090) were incubated for another 2 hours. The chemiluminescence detection kit (Thermo Fisher Scientific, Inc.) was used for visualization.
Immunofluorescence (IF) assay
H9C2 cells were fixed using 4% paraformaldehyde, and after blocking with 5% BSA and permeabilizing with 0.2% Triton X-100 in PBS, the cells were incubated with OLFM4 antibody (1 µg/mL; ab85046; Abcam, Shanghai, China). The cells were then incubated with FITC-labeled goat anti--rabbit IgG (Proteintech Group, Inc., Wuhan, China). DAPI was used for nuclear staining. IF images were obtained using the Olympus BX53 microscope (Olympus Optical Co. Ltd., Tokyo, Japan).
CCK-8 assay
H9C2 cells (1 × 104 cells/well) were cultured in a 96-well plate for 24 hours. Each well was then supplemented with 10 μL of Cell Counting Kit-8 (CCK-8) solution (Dojindo Laboratories, Kumamoto, Japan). After 4 hours, cell viability was determined by measuring absorbance at 450 nm using a spectrophotometer (Thermo Fisher Scientific, MA, USA).
5-ethynyl-2'-deoxyuridine (EdU) assay
H9C2 cells were incubated with EdU (50 μM, RiboBio, Guangzhou, China) solution for 2 h. After fixation with 4% paraformaldehyde and permeabilization with 0.5% Triton-X-100, the cells were stained with Apollo dye solution and 4',6-diamidino-2-phenylindole (DAPI) solution. EdU-positive cells (red) were observed under a fluorescence microscope (Leica, Hilden, Germany).
Flow cytometry
Cell apoptosis was assessed using the Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, Woburn, MA, USA). Briefly, the BrH9C2 cells were stained with FITC-labeled Annexin-V (5 μL) in the dark, followed by the addition of PI (10 μL). Cell apoptosis was then analyzed using a BD Accuri C6 Plus flow cytometer and the BD Accuri C6 Plus software (BD Biosciences, Franklin Lakes, NJ, USA).
ELISA
The levels of TNF-α, IL-6, and IL-1β in the supernatant were measured using ELISA kits for TNF-α (ab236712), IL-6 (ab234570), and IL-1β (ab255730) from Abcam (Shanghai, China).
Statistical analysis
Statistical analysis was performed using SPSS 22.0 software (IBM Corp., Armonk, NY, USA). The data are presented as the mean ± standard deviation (SD) and were normally distributed. Comparisons were made using Student's t-test (two-tailed unpaired) or one-way analysis of variance (ANOVA) followed by the Tukey post-hoc test. Each assay was repeated three times. A p-value of <0.05 was considered statistically significant.
Results
OLFM4 was highly expressed in LPS-stimulated H9C2 cells
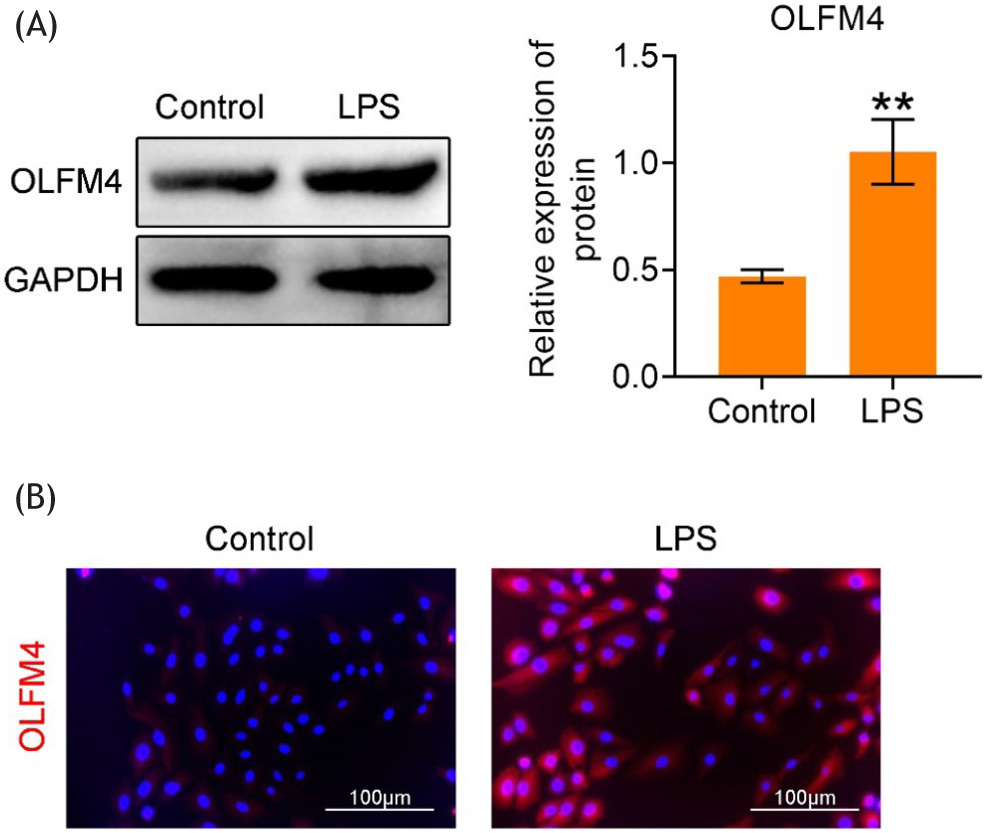
As shown in Figure 1A, OLFM4 protein expression was significantly upregulated in LPS-stimulated H9C2 cells (p<0.01). Immunofluorescence assay demonstrated that the fluorescence intensity of OLFM4 increased following LPS stimulation (Figure 1B). These findings indicate that OLFM4 is highly expressed in LPS-stimulated H9C2 cells.
Figure 1. Overexpression of OLFM4 in LPS-stimulated H9C2 cells. (A) Western blot analysis of OLFM4 protein expression in Control and LPS-treated groups. **p<0.01. (B) Immunofluorescence assay showing the fluorescence intensity of OLFM4 in Control and LPS-treated groups. **p<0.01.
Suppression of OLFM4 restrained cell apoptosis in LPS-triggered H9C2 cells
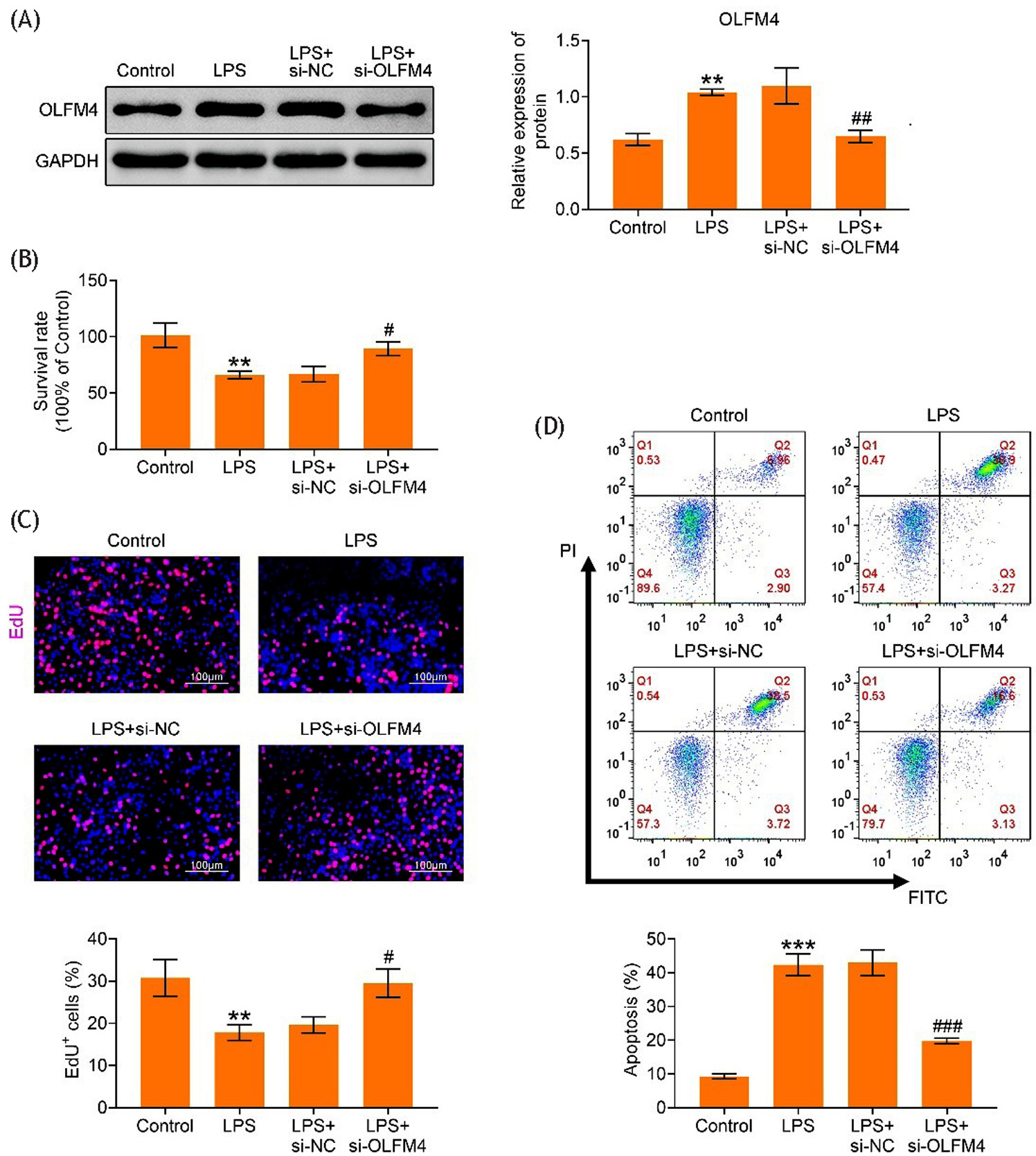
The knockdown efficiency of OLFM4 was confirmed based on the observed significant reduction in OLFM4 protein expression following OLFM4 suppression (p<0.01) (Figure 2A). Cell viability decreased in LPS-triggered H9C2 cells, but this reduction was mitigated after OLFM4 downregulation (p<0.05) (Figure 2B). Additionally, the number of EdU-positive cells decreased following LPS treatment, an effect reversed by OLFM4 inhibition (p<0.05) (Figure 2C). Cell apoptosis increased in LPS-triggered H9C2 cells, but this effect was significantly attenuated following OLFM4 knockdown (p<0.001) (Figure 2D). Overall, suppression of OLFM4 reduced cell apoptosis in LPS-triggered H9C2 cells.
Figure 2. Suppression of OLFM4 restrained cell apoptosis in LPS-triggered H9C2 cells. The groups were divided into Control, LPS, LPS+si-NC, and LPS+si-OLFM4. (A) Western blot analysis of OLFM4 protein expression in different groups. (B) Cell survival rate assessed by CCK-8 assay. (C) Cell proliferation evaluated using the EdU assay. (D) Cell apoptosis determined by flow cytometry. **p<0.01, ***p<0.001 vs. control group; #p<0.05, ##p<0.01, ###p<0.001 vs. LPS+si-OLFM4 group.
Knockdown of OLFM4 alleviated inflammation in LPS-mediated H9C2 cells
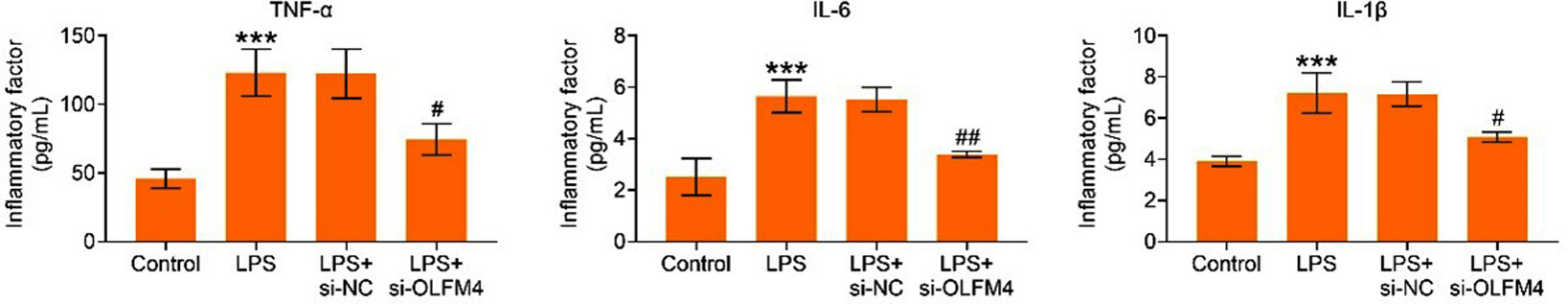
The levels of inflammatory factors TNF-α, IL-6, and IL-1β were elevated following LPS treatment, and these increases were significantly reduced after silencing OLFM4 (p<0.05) (Figure 3), suggesting that the knockdown of OLFM4 alleviates inflammation in LPS-mediated H9C2 cells.
Figure 3. Knockdown of OLFM4 alleviated inflammation in LPS-mediated H9C2 cells. The groups were divided into Control, LPS, LPS+si-NC, and LPS+si-OLFM4. The levels of inflammatory factors TNF-α, IL-6, and IL-1β were assessed using ELISA. ***p<0.001 vs. control group; #p<0.05, ##p<0.01 vs. LPS+si-OLFM4 group.
Inhibition of OLFM4 retarded the NF-κB signaling pathway
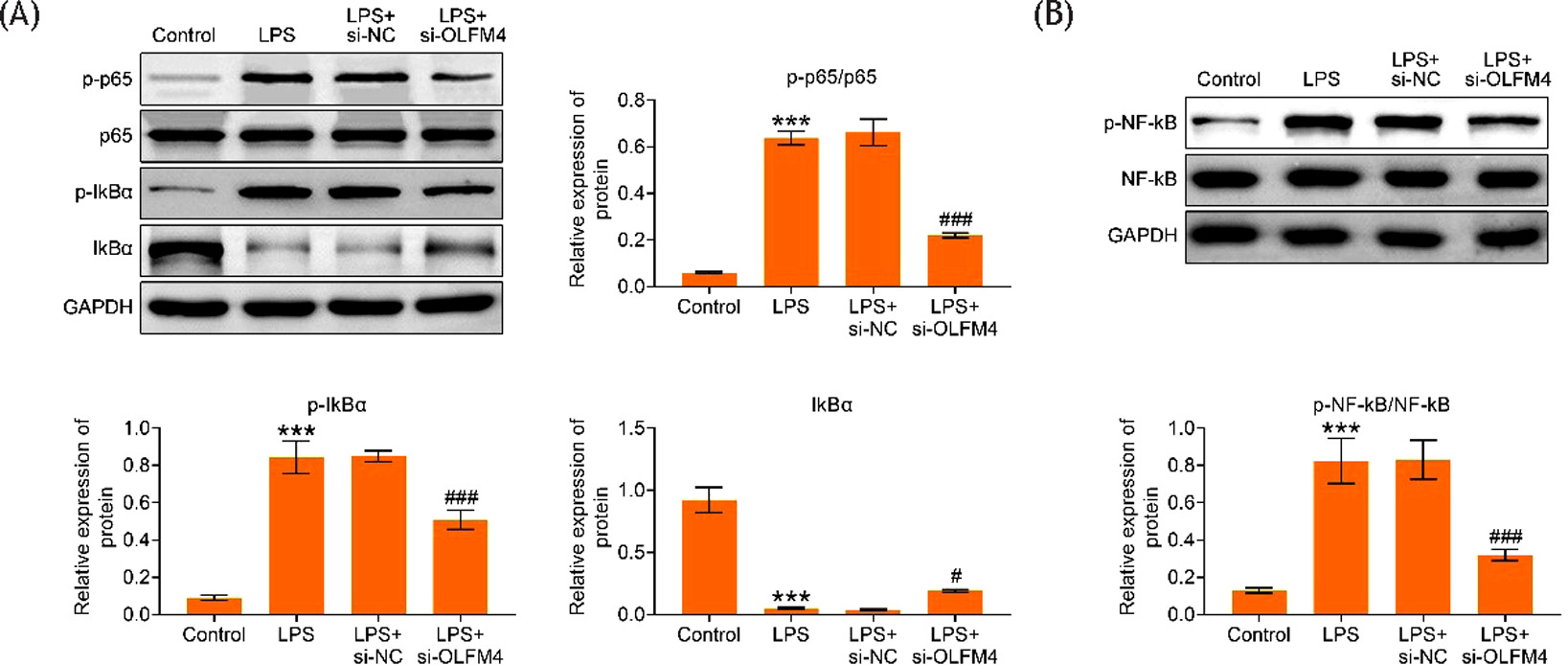
Next, the protein expressions of p-p65/p65 and p-IkBα were found to be elevated, and IkBα expression was reduced in LPS-mediated H9C2 cells. However, these changes were counteracted by OLFM4 inhibition (p<0.05) (Figure 4A). In addition, the protein expression of p-NF-κB/NF-κB was increased in LPS-triggered H9C2 cells, but this effect was offset following OLFM4 suppression (p<0.001) (Figure 4B). In summary, inhibition of OLFM4 retarded the NF-κB signaling pathway.
Figure 4. Inhibition of OLFM4 suppressed the NF-κB signaling pathway. The groups were divided into Control, LPS, LPS+si-NC, and LPS+si-OLFM4. (A) Western blot analysis of the protein expressions of p-p65, p65, p-IkBα, and IkBα. (B) Western blot analysis of the protein expressions of p-NF-κB and NF-κB. ***p<0.001 vs control group; #p<0.05, ###p<0.001 vs. LPS+si-OLFM4 group.
Discussion
This study demonstrated that OLFM4 protein expression was significantly elevated in LPS-stimulated H9C2 cells, and suppression of OLFM4 enhanced cell proliferation and inhibited cell apoptosis in LPS-triggered H9C2 cells. The levels of inflammatory factors TNF-α, IL-6, and IL-1β were elevated following LPS treatment, but these increases were mitigated by silencing OLFM4. Additionally, inhibition of OLFM4 was shown to retard the NF-κB signaling pathway.
LPS has been used to establish a cell model for sepsis due to its ability to activate inflammatory responses.14 In this study, H9C2 cells were treated with LPS to mimic a septic myocardial injury model. Various proteins have been investigated for their roles in regulating sepsis-induced myocardial injury. Notably, a previous report identified OLFM4 as a critical gene in sepsis through bioinformatics analysis.13 However, the regulatory functions and associated pathways of OLFM4 in sepsis-induced myocardial injury were previously unknown. Our present study demonstrated that OLFM4 protein expression was significantly elevated in LPS-stimulated H9C2 cells. Furthermore, suppression of OLFM4 enhanced cell proliferation and inhibited cell apoptosis in LPS-triggered H9C2 cells.
Inflammation is a pivotal process in sepsis-induced myocardial injury, and many researchers have focused on modulating inflammation in this context. For example, Hsp22 has been shown to reduce inflammation and oxidative stress to ameliorate LPS-induced myocardial injury.15 Additionally, ulinastatin inhibits NLRP3 inflammasome activation to attenuate sepsis-induced myocardial injury.16 Exogenous fetuin-A alleviates oxidative stress and inflammation to protect against sepsis-induced myocardial injury in mice.17 Furthermore, in sepsis-mediated myocardial injury, miR-195-5p modulates ATF6 to mitigate inflammation and endoplasmic reticulum stress.18 Similar to these previous studies, our present study demonstrated that the levels of inflammatory factors TNF-α, IL-6, and IL-1β were elevated following LPS treatment, but these increases were mitigated by silencing OLFM4.
The NF-κB signaling pathway plays an essential role in sepsis-induced myocardial injury. For instance, neogambogic acid modulates the p38 MAPK/NF-κB pathway to ameliorate sepsis-induced myocardial injury.19 Additionally, in septic rats, rosiglitazone regulates the NF-κB pathway to improve myocardial injury.20 Suppression of XBP1 inhibits the NF-κB signaling pathway to relieve LPS-induced cardiomyocyte injury.21 Notably, OLFM4 has been shown to affect the NF-κB signaling pathway in sepsis.12 Consistent with these studies, our results demonstrated that inhibition of OLFM4 retarded the NF-κB signaling pathway.
Although our study's findings provide interesting insights into potentially improving the clinical treatment of sepsis-induced myocardial injury, it had some limitations, such as the lack of investigations into other phenotypic aspects (e.g., autophagy, oxidative stress, mitochondrial injury, and ferroptosis), as well as the absence of animal experiments and clinical investigations. Future studies could further explore the role of OLFM4 in sepsis-induced myocardial injury.
Conclusion
The knockdown of OLFM4 protected cardiomyocytes from sepsis by inhibiting apoptosis and inflammatory responses via the NF-κB pathway, suggesting that OLFM4 could be a potential therapeutic target for treating sepsis-induced myocardial injury. Further studies are needed to explore these findings' broader implications and potential clinical applications.
Conflict of Interest
The authors declare no conflict of interest.
Consent to participate statement
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Data availability
The authors declare that all data supporting this study's findings are available within the paper, and any raw data can be obtained from the corresponding author upon request.
Author Contributions
Hailu Chen and Guihua Fang designed and carried out the study. Hailu Chen and Shuna Liu supervised the data collection, analyzed the data, and interpreted the data. Hailu Chen and Guihua Fang prepared the manuscript for publication and reviewed the draft. All authors have read and approved the manuscript.
REFERENCES
1. Ackerman, MH, et al., Sepsis. Crit Care Nurs Clin North Am. 2021;33(4):407-18. 10.1016/j.cnc.2021.08.003
2. Sazonov, V, et al. Case Series: Efficacy and safety of hemoadsorption with ha-330 adsorber in septic pediatric patients with cancer. Front Pediatr. 2021;9:672260. 10.3389/fped.2021.672260
3. Zuccari, S, et al. Changes in cytokines, haemodynamics and microcirculation in patients with sepsis/septic shock undergoing continuous renal replacement therapy and blood purification with cytosorb. Blood Purif. 2020;49(1-2):107-13. 10.1159/000502540
4. Mirijello, A, Tosoni, A, On Behalf Of The Internal Medicine Sepsis Study Group. New strategies for treatment of-sepsis. Medicina. 2020;56(10):527. 10.3390/medicina56100527
5. Carbone, F, et al. Septic cardiomyopathy: from pathophysiology to the clinical setting. Cells. 2022;11(18). 10.3390/cells11182833
6. Hochstadt, A, Meroz Y, Landesberg G. Myocardial dysfunction in severe sepsis and septic shock: more questions than answers? J Cardiothorac Vasc Anesth, 2011;25(3): 526-35. 10.1053/j.jvca.2010.11.026
7. Liu, W, Rodgers, GP. Olfactomedin 4 Is a biomarker for the severity of infectious diseases. Open Forum Infect Dis. 2022;9(4):ofac061. 10.1093/ofid/ofac061
8. Chen, S, et al. Olfactomedin 4 deletion exacerbates nonalcoholic fatty liver disease through P62-dependent mitophagy in mice. Metabolism. 2023;148:155679. 10.1016/j.metabol.2023.155679
9. Ma, HW, et al. Olfactomedin 4 produces dysplasia but suppresses metastasis of colon cancer. Cancer Gene Ther. 2023;30(5):694-703. 10.1038/s41417-022-00585-9
10. Ren, X, et al. Quantitative proteomic analysis of synovial tissue reveals that upregulated OLFM4 aggravates inflammation in rheumatoid arthritis. J Proteome Res. 2021;20(10):4746-57. 10.1021/acs.jproteome.1c00399
11. Zhang, F, et al. Qinbaohong Zhike oral liquid attenuates LPS-induced acute lung injury in immature rats by inhibiting OLFM4. Oxid Med Cell Longev. 2022;2022:7272371. 10.1155/2022/7272371
12. Gong, F, et al. OLFM4 regulates lung epithelial cell function in sepsis-associated ARDS/ALI via LDHA-mediated NF-κB signaling. J Inflamm Res. 2021;14:7035-51. 10.2147/JIR.S335915
13. Abdelnaser, M, et al. Modulating Nrf-2/HO-1, apoptosis and oxidative stress signaling pathways by gabapentin ameliorates sepsis-induced acute kidney injury. Naunyn Schmiedebergs Arch Pharmacol, 2024;397(2):947-58. 10.1007/s00210-023-02650-y
14. Fang, X. et al., The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol, 2023;20(1):7-23. 10.1038/s41569-022-00735-4
15. Yu, Y. et al., Hsp22 ameliorates lipopolysaccharide-induced myocardial injury by inhibiting inflammation, oxidative stress, and apoptosis. Bioengineered. 2021;12(2):12544-54. 10.1080/21655979.2021.2010315
16. Qiu, J, et al. Ulinastatin protects against sepsis-induced myocardial injury by inhibiting NLRP3 inflammasome activation. Mol Med Rep. 2021;24(4). 10.1002/iid3.1039
17. Sidheeque Hassan, V, et al. Exogenous fetuin-A protects against sepsis-induced myocardial injury by inhibiting oxidative stress and inflammation in mice. Fundam Clin Pharmacol. 2023;37(3):607-17. 10.1111/fcp.12870
18. Xia, H, et al. MiR-195-5p represses inflammation, apoptosis, oxidative stress, and endoplasmic reticulum stress in sepsis--induced myocardial injury by targeting activating transcription factor 6. Cell Biol Int. 2022;46(2):243-54. 10.1002/cbin.11726
19. Fu, W, et al. Neogambogic acid relieves myocardial injury induced by sepsis via p38 MAPK/NF-κB pathway. Korean J Physiol Pharmacol. 2022;26(6):511-18. 10.4196/kjpp.2022.26.6.511
20. Zhang, SM, et al. Effect of rosiglitazone on myocardial injury in septic rats through NF-κB pathway. Eur Rev Med Pharmacol Sci. 2020;24(1):452-60. 10.26355/eurrev_202001_19945
21. Zhang, C, et al. Inhibition of XBP1 Alleviates LPS-Induced Cardiomyocytes Injury by Upregulating XIAP through Suppressing the NF-κB Signaling Pathway. Inflammation, 2021;44(3):974-84. 10.1007/s10753-020-01392